
Il progetto finanziato dal bando AIRett 2016 si inserisce nel contesto della linea di ricerca volta ad
identificare terapie che possano essere o risolutive o migliorative per i sintomi della sindrome di Rett. Pensando ad una strategia collettiva in cui più gruppi di ricerca indirizzano i loro sforzi in più direzioni per un comune obiettivo, il nostro lavoro si colloca nella linea di studio che si adopera per creare dei modelli che siano il più possibile vicino alla situazione reale delle bambine Rett e che rappresentino un valido strumento per verificare l’efficacia di nuovi trattamenti, sia farmacologici, sia basati su principi più nuovi.
La maggior parte degli studi condotti fino ad oggi sulla sindrome di Rett infatti hanno utilizzato il modello animale, prevalentemente murino, sia per lo studio sia per la valutazione dell’efficacia di nuove molecole. Sono stati generati più modelli di topi transgenici, sia di sesso maschile che femminile, ma solo dal 2016 sono stati creati anche animali con mutazioni presenti nelle bambine (ref). Restando fondamentale l’importanza del modello animale che permette di studiare l’organismo in toto e le sue risposte, la complessità del cervello umano è molto superiore a quella murina, così come la complessità del genoma e dell’epigenoma, fattori che influenzano sia l’andamento della malattia, sia la possibile cura. A tale scopo da diversi anni, è stata sviluppata una tecnologia che consente di trasformare cellule differenziate di un organismo in cellule staminali, più precisamente cellule staminali pluripotenti indotte, IPSCs, che hanno come le cellule staminali la potenzialità di differenziarsi in tipi cellulari specifici. Tali cellule costituiscono oggi in medicina un’importante risorsa per lo studio della malattia, ma non solo, infatti sono già utilizzate anche per medicina rigenerativa.
Le prime IPSC derivate da pazienti con lasindromedi Rett risalgono al 2010 (Marchetto et al); successivamente altri gruppi hanno generato IPSCs da pazienti con mutazioni nel gene MECP2, a partire da fibroblasti mediante l’utilizzo di virus che si integrano nel genoma ed inducono la cellula ad esprimere fattori di staminalità. Tuttavia solo alcune delle mutazioni più frequenti nelle pazienti con sindrome di Rett, sono state generate; per molte mutazioni “hot spot” non esiste un modello paziente specifico. Allo scopo di creare un modello cellulare il più possibile simile alle reali cellule della paziente nel tempo le tecnologie si sono affinate ed oggi è possibile creare modelli cellulari con sistemi non integrativi, ed a partire da tessuti molto accessibili, quali ad esempio il sangue. Cosa si intende con sistemi non integrativi? Significa che i vettori che inseriamo nelle cellule, negli eritroblasti del sangue nel nostro caso, non si integrano nel genoma, ma vengono eliminati in poche ore, e non modificano o comunque solo in maniera transitoria il comportamento naturale della cellule.
Un’altra questione molto importante, quando si usa un sistema modello è la disponibilità di un controllo sano che permetta al ricercatore di valutare quali siano le differenze tra la cellula che porta il difetto genetico e la cellula “sana”. Poiché il gene MECP2 ed il gene CDKL5 mappano sul cromosoma X e poiché nelle donne in ogni cellula solo uno dei due cromosomi X è attivo, nelle bambine in cui questo fenomeno interessa il 50% delle cellule si può ottenere da ogni paziente cloni “sani” da cui si genereranno tipi cellulari differenziati che costituiscono il controllo sano (cellule neuronali, ma non solo ad es. anche cardiomiociti, di interesse per le pazienti con compromissione cardiologica), e cloni che esprimono la mutazione, rappresentativi della sindrome di Rett. Questo confronto tra cellule mutate e cellule sane di uno stesso individuo (controllo ISOGENICO) è ottimale, perché ci consente di valutare differenze che sono funzione del solo difetto genetico di MECP2 o CDKL5 senza le influenze derivate dal diverso genotipo, come invece accade quando si confrontano le cellule del paziente con quelle di individui sani non correlati. La disponibilità di un adeguato controllo per ogni paziente è un’indispensabile punto di partenza per l’arduo compito di identificare marcatori biologici della malattia che, nei tipi cellulari di nostro interesse rivelino differenze, se possibili misurabili, tra cellula sana e malata, di cui verificare la reversibilità in seguito al trattamento.
La novità dello studio da noi intrapreso consiste quindi nella selezione di casi con mutazioni “hot spot” per cui ancora non sono state generate IPSCs ed in particolare abbiamo scelto mutazioni associate a quadri clinici di diversa severità, quali p.Arg168* (generalmente la manifestazione clinica è severa), p.Arg255(associata ad una manifestazione clinica moderata) e p.Arg133Cys, associata a pazienti con un quadro clinico lieve. Attualmente abbiamo reclutato un paio di pazienti per mutazione ma in entrambe le pazienti con mutazione p.Arg 168 ed in una paziente pArg255* l’inattivazione del cromosoma X appariva sbilanciata (vedi tabella a fianco).
L’intero processo di differenziamento neuronale richiede una prima fase in cui si producono cloni di cellule staminali, una seconda fase di induzione neuronale, ed infine il differenziamento finale nei tipi di neuroni desiderati, corticali ed ippocampali.
A questo progetto lavorano attivamente oltre alla dott.ssa Russo, la dott.ssa Valentina Alari, la dottssa Francesca Cogliati. La caratterizzazione viene eseguita dalle dott. sse Valentina Giorgini, Ilaria Catusi e Maria Garzo (Figura 1).
Ad oggi è stato possibile ottenere cloni IPCs per le 2 pazienti con mutazione p.Arg133Cys. Sono stati selezionati 14 cloni per ciascuna paziente, valutata sul cDNA l’espressione del gene MECP2 in 8 di questi, in modo da disporre di almeno tre cloni dell’allele mutato, e tre dell’allele sano per ciascuna paziente (figura 2).
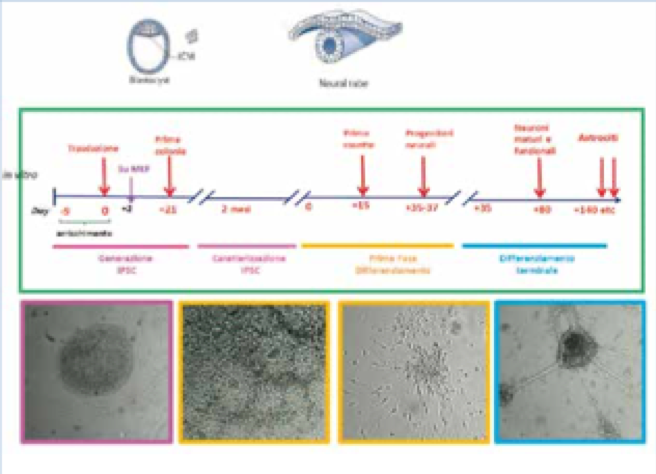
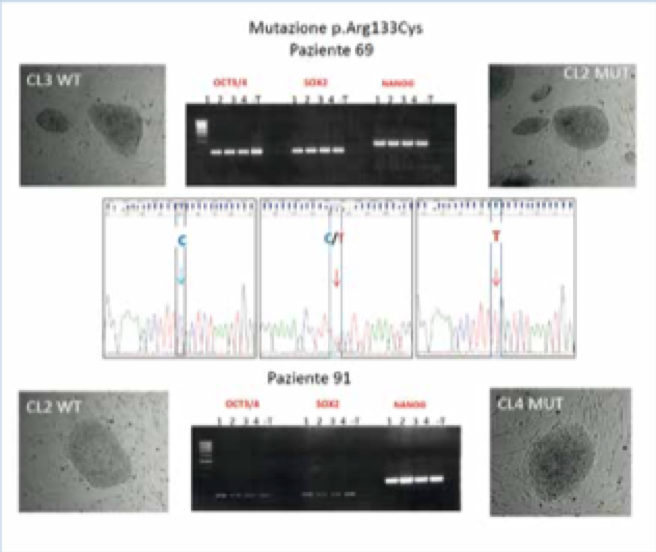
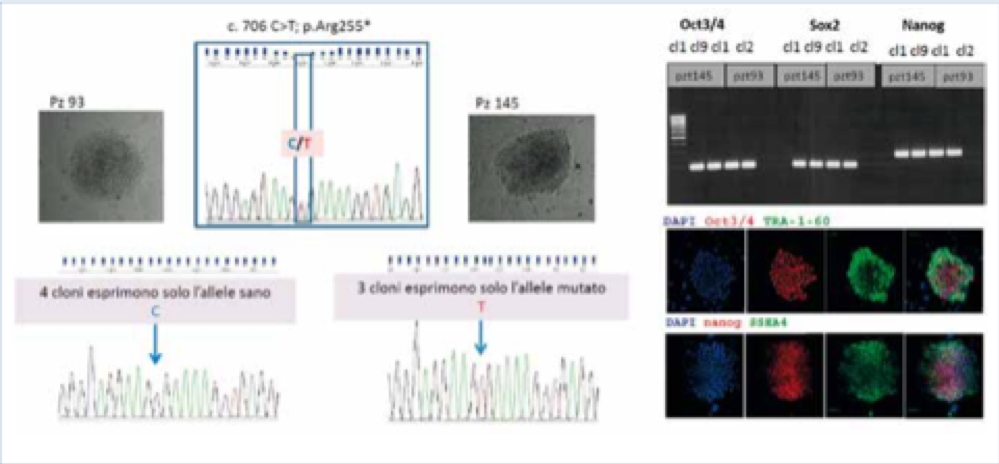
Invece, per le pazienti che esprimono la mutazione p.Arg255, abbiamo selezionato 3 cloni che esprimono l’allele mutato di una paziente e 5 cloni che esprimono l’allele sano dell’altra (Figura 3). In seguito al risultato è stata rivalutata l’inattivazione preferenziale del cromosoma X, che si è rivelata correttamente bilanciata nella paziente 145, ove procederemo ad una successiva riprogrammazione per generare cloni di controllo, mentre nella paziente 93 l’inattivazione nel sangue è apparentemente sbilanciata, quindi la maggior parte delle cellule del sangue esprime un allele sano. Una volti scelti i cloni delle cellule staminali utili al nostro studio, bisogna confermare che le cellule siano effettivamente staminali. Ciò viene fatto valutando l’espressione di marcatori di staminalità come indicato nelle figure, sia mediante RT PCR dei geni OCT SOX e NANOG sia con tecniche di immunofluorescenza che verificano anche l’espressione delle proteine TRA1-60 e SSEA4. Infine prima di poter differenziare i cloni selezionati in neuroni, occorre verificare che durante il processo di generazione delle IPSCs la mappa cromosomica sia rimasta fedele all’originale, ossia che non siano insorti riarrangiamenti prima non presenti nel paziente che potrebbero alterare l’interpretazione di marcatori biologici e quindi dell’efficacia di molecole testate su di esse. A tale scopo viene effettuata la mappa cromosomica sia del sangue della bambina sia dei cloni di IPSC derivato da questo. Abbiamo eseguito per ora questo passaggio sulle IPScs delle pazienti con mutazione p.Arg255 ed è in corso per quelle delle pazienti p.Arg133Cys.
A questo punto siamo pronti per entrare nel vivo del lavoro differenziando le cellule in neuroni corticali e ippocampali. Alcune aliquote delle cellule staminali generate verranno conservate per esperimenti futuri e rese eventualmente disponibili, con il consenso dei genitori per progetti di collaborazione internazionale volti a saggiare strategie terapeutiche. Sarebbe auspicabile reclutare qualche paziente in più con una di queste tre mutazioni, in particolare bambine con un quadro clinico piuttosto severo.
