
Speciale Congresso Lido di Camaiore 2007
Dott. ssa Silvia Russo – Istituto Auxologico Italiano Milano
La sindrome di Rett è una malattia che interessa quasi esclusivamente bambine ed è conseguente alla presenza di mutazioni nel gene MethylCpG-binding Protein 2, MeCP2 in almeno l’80% delle pazienti. Il gene MeCp2 è localizzato sul cromosoma X e nelle pazienti uno solo dei due cromosomi porta il difetto genetico che nel 99,5% insorge “de novo”, ossia nessuno dei genitori ne è portatore. Solo in una piccolissima percentuale di donne, lo 0,5%, è stata riscontrata la mutazione in assenza di malattia: questo può avvenire in quanto, per un meccanismo noto come inattivazione selettiva del cromosoma X, il cromosoma che porta la mutazione viene preferenzialmente inattivato (contrariamente a quanto avviene di solito con un’inattivazione casuale del 50% di ciascun cromosoma X) e quindi le conseguenze della mutazione non si manifestano. La presenza di un solo cromosoma X nei maschi aveva inizialmente indotto a pensare che, in essi, le mutazioni nel gene MeCP2 non potessero essere compatibili con la vita. In realtà anche in un piccolo numero di individui di sesso maschile sono state riscontrate mutazioni in questo gene con un quadro clinico si differenzia da quello osservato nelle bambine. Sulla base del tipo di mutazione e presentazione clinica si possono distinguere tre diversi gruppi di pazienti maschi con mutazione nel gene MeCP2:
- pazienti con con encefalopatia precoce severa, portatori di mutazioni riscontrate anche in bambine con fenotipo classico (Thr158Met, Gly163fs,Gly252fs, Gly269fs) e Pro385fs riscontrata in una bambina non affetta con uno sbilanciamento selettivo del cromosoma X;
- pazienti con fenotipo Rett-like e mutazioni di MeCP2 sia già osservate in bambine con Rett classica, (Arg133Cys, Ser134Cys, Tyr141XThr158Met, Arg270X), sia mai riportate (Gly273fs, Arg344Trp) in condizione di mosaicismo conseguente ad una condizione di aneuploidia cromosomica dell’X ad esempio una sindrome di Klinefelter. In altri casi si trattava di mosaicismo somatico sempre per mutazioni sia descriite in associazione di un quadro classico (Arg133His, Arg270X) sia mai riportate (Pro56fs). Tra le mutazioni riportate in questo secondo gruppo solo quelle che alterano l’Arg133 sono state descritte in ragazze RTT con forme clinicamente meno severe.
- maschi con ritardo mentale lieve/moderato, portatori di mutazioni ereditate dalle loro madri sane e non descritte nelle pazienti RTT (Glu137Gly, Ala140Val, Arg167Trp, Ala181Val, Pro225Leu, Thr228Ser, Lys284Glu, Pro387Leu, Pro387-Met466del80, Gln406X, Arg453Gln, Arg471fs, Glu472fs). In questo caso nessuna delle mutazioni è mai stata osservata in femmine con la sindrome di Rett, l’inattivazione del cromosoma X nelle madri portatrici, dove è stata valutata, risultava correttamente bilanciata. Unica eccezione per la mutazione Pro387-Met466del80 dove la mamma del paziente aveva un’inattivazione selettiva dell’X.
Presso i laboratori di Genetica Molecolare dell’Istituto Auxologico Italiano (Mi) è attivo un servizio di diagnostica molecolare e di ricerca sulla sindrome di Rett. Più di 300 bambine con sospetta sindrome di Rett e oltre 50 maschi sono stati valutati per la presenza di mutazioni nel gene MeCP2; all’interno di quest’ultimo campione è stato identificato un bambino con mutazione nella porzione carbossiterminale di MeCP2 e precisamente una delezione di 44bp, c.1163_1207del44bp, p.Pro389X.
La condizione clinica del bambino come intermedia tra una encefalopatia severa e Rett-like. Successivamente ad una gravidanza normodecorsa, a parto e periodo perinatale nella norma, si è definito un quadro di grave encefalopatia, con presenza di un’epilessia precoce caratterizzata da crisi farmacoresistenti e con tracciato EEG caratterizzato da parossismi epilettici a distribuzione multifocale. E’ riportata una regressione delle capacità motorie, da sempre assenza di linguaggio, microcefalia, assenza di presa nella mani e di stereotipie, presenza di apnee minime. La mutazione risultava ereditata per via materna e lo studio dell’inattivazione del cromosoma X ha evidenziato un valore sufficiente per un’inattivazione selettiva. Prima dell’analisi di MeCP2 erano stati eseguiti parecchi test, tra cui la mappa cromosomica che ha verificato l’occorrenza di un cariotipo 46X,Y escludendo così aneuploidie del cromosoma X. L’erediteriatà materna della mutazione esclude la presenza di mosaicismo somatico. Lo studio del cDNA del paziente ha evidenziato la presenza del solo trascritto mutato, che non risultando degradato produrrà quindi una proteina anomala.
La figura riportata sotto mostra gli elettroferogrammi di un soggetto normale, del paziente a cui mancano 44 nucleotidi e della mamma che presenta un gene normale ed uno con la delezione.
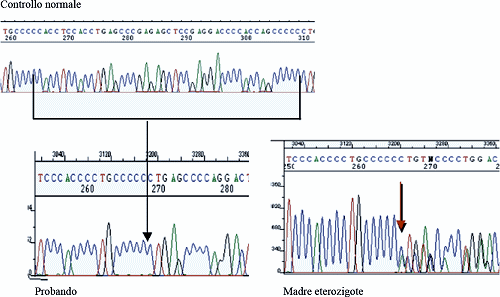
Il riscontro di questi rari pazienti emizigoti con mutazione del gene MeCP2 costituiscono uno strumento molto importante per la ricerca, perché permettono di studiare le conseguenze della mutazione in assenza del cromosoma X sano presente invece nelle bambine.
La mutazione P389X non era mai stata descritta in maschi con un quadro un clinico severo, ma era già stata osservata in bambine Rett. Una revisione della nostra casistica di pazienti mutate e, successivamente, della letteratura ha evidenziato come questa mutazione apparisse legata al quadro clinico più benigno delle varianti a linguaggio preservato. Tutte e 4 le bambine da noi diagnosticate con questa mutazione presentavano, a differenza della madre del probando, un’inattivazione bilanciata del cromosoma X, vale a dire esprimevano il 50% del gene con mutazione ed il 50% del gene normale.
Tre di queste 4 pazienti, a noi inviate da reparti di Neuropsichiatria Infantile sul territorio, sono state riviste dalla dott.ssa Bonati che ha valutato in modo specifico l’aspetto comportamentale e verificato la presenza di linguaggio preservato. Le informazioni forniteci dai colleghi che hanno in cura le bambine unite alla valutazioni fatte presso il nostro istituto sembrano definire un quadro simile per queste pazienti che si differenziano dalle Rett classiche non solo per la presenza di un linguaggio preservato. Solo una delle bambine ha manifestato una vera e propria regressione, che si è manifestata molto tardivamente (30 mesi), nessuna ha perso l’uso finalizzato delle mani tutte camminano senza aiuto; presentano tutte epilessia seppure in forma lieve e controllata, apnee minime e rientrano nello spettro dell’autismo considerando il periodo di vita attorno ai 4-5 anni.
Per tutte le pazienti e per il maschio con la stessa mutazione sono state allestite linee linfoblastoidi. Sarà oggetto di studio nei prossimi mesi il confronto tra il profilo di espressione del maschio affetto, delle pazienti femmine con la stessa mutazione, di pazienti con mutazioni di stop precoci e di controlli normali maschi e femmine. Lo studio sarà eseguito utilizzando la piattaforma Illumina con il modulo Gene Expression.
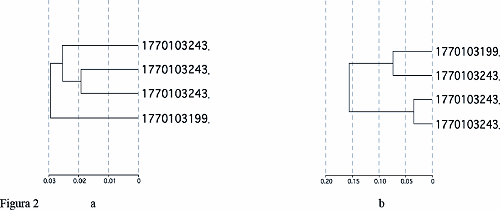
Un primo esperimento condotto su una bambina con PVS e sul maschio con la stessa mutazione a confronto con un soggetto maschio ed una femmina normale hanno evidenziato la presenza di circa 500 differenzialmente espressi. Il risultato interessante è che se confrontiamo tutti e 4 i soggetti per tutti i geni (48000) non si notano particolari associazioni (figura 2a), ma se li confrontiamo per i soli geni differenzialmente espressi notiamo che il maschio e la femmina mutati sono più simili tra loro, ed analogamente i non mutati (figura 2b). Ciò ci incoraggia a continuare i nostri studi su questo tessuto che pur non essendo il tessuto nervoso, dove la proteina svolge il suo ruolo principale, contiene geni che potrebbero essere regolati da MeCP2 ed avere un ruolo nell’espressione del quadro clinico.
Questo lavoro è frutto della collaborazione di:
- Silvia Russo
- Maria Teresa Bonati
- Francesca Cogliati
- Maura Masciadri
- Lidia Larizza
- Istituto Auxologico Italiano Milano
- Dott.ssa Isabella Moroni
- Dott.ssa Elena Freri
- Istituto Neurologico Carlo Besta- Milano
- Dott. Pierangelo Veggiotti
- Istituto Mondino – Pavia
- Dott Lucio Giordano
- Spedali Civili Brescia
