
Presentiamo in queste pagine i primi risultati dello studio sull’epilessia introdotto nell’articolo precedente.
Maria Pintaudi – Istituto G. Gaslini (Università di Genova), Istituto San Paolo (Milano)
Le crisi epilettiche, la loro imprevedibilità, frequenza e gravità, hanno un impatto importante sulla qualità della vita di tutte le persone con epilessia, ma particolarmente in condizioni come la Sindrome di Rett (SR), dove ci sono anche altre importanti comorbilità.
Nonostante l’epilessia sia presente in circa l’80% delle pazienti con SR, gli studi clinici che indagano le caratteristiche delle crisi (tipologia, età di esordio, evoluzione, correlazione con altri fattori, compresi quelli genetici) e l’uso e l’efficacia dei farmaci anti-epilettici sono scarsi.
Recentemente è stato pubblicato uno studio australiano (Le Jan et al 2006) di 162 pazienti affetti da SR che evidenzia come le crisi tendano ad essere peggiori in soggetti che presentano una compromissione maggiore dello stato clinico generale e che mutazioni come 294X e R255X e delezioni C terminali sembrino essere associate ad una frequenza minore di epilessia.
 Maria Pintaudi
Maria Pintaudi
Per quanto riguarda il trattamento delle crisi sono attualmente disponibili solo pochi dati. Carbamazepina, Valproato di sodio, Sulthiame e Lamotrigina sembrano essere i farmaci anti-epilettici più frequentemente utilizzati. Tuttavia gli studi attualmente a disposizione sono di piccole dimensioni e non ci sono ad oggi lavori che confrontino l’efficacia dei vari farmaci antiepilettici nella Sindrome di Rett. Ne consegue quindi che la scelta di utilizzare un farmaco è attualmente basata sull’esperienza individuale e sulla preferenza del medico che ha in carico il paziente.
Data la carenza di studi, su proposta dell’AIR, Associazione Italiana Sindrome di Rett, è nato quindi un progetto di studio sull’epilessia nella Sindrome di Rett.
Lo scopo è quello di:
- studiare la clinica dell’epilessia nella SR, le correlazioni con l’età, con il genotipo, con le caratteristiche cliniche;
- individuare gli approcci terapeutici più efficaci.
E’ stato quindi preparato un questionario per ogni paziente in cui viene chiesto di ricostruire, per quanto possibile, la storia epilettica del soggetto: età di esordio delle crisi, tipo di crisi, reperti EEG, farmaci utilizzati ed eventuale beneficio. Questo questionario viene proposto al medico che ha in carico il paziente.
Lo studio vede attualmente la partecipazione dei seguenti istituti:
- Istituto G. Gaslini (Università di Genova) Centro epilessia e Centro di riferimento regionale SR U.O e cattedra di Neuropsichiatria Infantile Responsabile: Prof E. Veneselli. Referente: dott. M. Pintaudi.
- Istituto San Paolo (MI) Centro epilessia e Centro di riferimento regionale SR. Responsabile Prof. M.P. Canevini. Referente: dott. A. Vignoli.
- Azienda Ospedaliera Fatebenefratelli (MI) Centro epilessia. Neurologia Pediatrica Responsabile e referente: Dott. Antonino Romeo.
- Spedali Civili di Brescia, Neuropsichiatria Infantile. Centro epilessia. Referente: Dott. Giordano.
- Azienda Ospedaliera Senese. Policlinico “Le Scotte”. Neuropsichiatria Infantile. Responsabile: Dott. J. Hayek.
- Ospedale Pediatrico Meyer, Università di Firenze. Clinica di Neurologia pediatrica. Responsabile: Prof . R. Guerrini.
Al Convegno Nazionale tenutosi a Rimini il 17 e 18 maggio abbiamo presentato i dati preliminari relativi alla casistica dell’Istituto G. Gaslini (Genova), dell’Istituto San Paolo (Milano) e degli Spedali Civili (Brescia).
In tabella 1 sono riportati i dati clinici generali.
Tabella 1
| N° pazienti |
50 |
| Età |
Range: 18 ms-34 anni (media 13 anni) |
| Forme Classiche |
39 (78%) |
| Forme Varianti |
11 (22%) (4 Hanefeld, 3 Preserved Speech, 4 Fruste) |
| MeCP2 + |
38 (76%) |
| CDKL5 + |
3 (6%) |
| Negative all’indagine genetica |
9 (18%) |
In tabella 2 sono riportati i dati relativi alla presenza di epilessia nelle pazienti SR.
Tabella 2
| Pz. epilettiche totali |
40 (80%) |
| Pz. epilettiche MecP2 + |
33 (87%) |
| Pz epilettiche CDKL5 + |
3 (100%) |
| Pz epilettiche MecP2 e CDKL5 – |
4 (44%) |
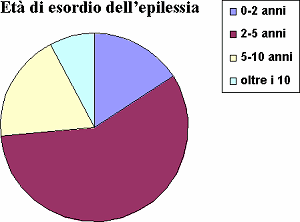
Età di esordio dell’epilessia
La nostra casistica attuale risulta sovrapponibile a quelle della letteratura per l’età (la maggior parte delle pazienti sono in stadio 3 o 4) e per l’età di esordio dell’epilessia, che corrisponde, in media, a 4.5 anni.
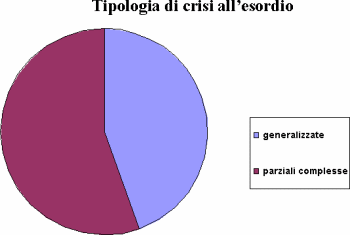
Tipologia di crisi all’esordio
Le crisi all’esordio sono parziali complesse (56%) e generalizzate (44%) di tipo tonico, tonico-clonico, mioclonico, atonico, assenze.
E’ in corso l’elaborazione dei dati concernenti l’inquadramento nosologico secondo la Classificazione Internazionale delle Epilessie e delle Sindromi epilettiche e la sua variazione nel tempo.
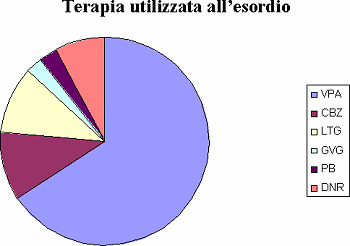
Terapia utilizzata all’esordio
Tutte le pazienti hanno iniziato con una monoterapia (Vedere tabella 2)
Tabella 2
| Farmaci utilizzati all’esordio |
Casi e % |
| VPA |
25 (66%) |
| CBZ |
4 (11%) |
| LTG |
4 (11%) |
| GVG |
1 (2%) |
| PB |
1 (2%) |
| DNR |
3 (8%) |
 Legenda
Legenda
(VPA: valproato di sodio, CBZ: carbamazepina,
LTG: lamotrigina, GVG: vigabatrin,
PB: fenobaribital, DNR: dato non reperibile)
L’efficacia della terapia all’esordio è descritta nella tabella 3.
Tabella 3
| Efficacia | 0 | 1 | 2 | Non nota |
| VPA | – | 12 (48%) | 11(44%) | 2 (8%) |
| CBZ | – | 2 (50%) | 2 (50%) | – |
| LTG | – | 2 (50%) | 2 (50%) | – |
| GVG | – | 1 | – | – |
| PB | – | 1 | – | – |
Legenda
0: nessun effetto 1: riduzione frequenza crisi ? 50%
2: scomparsa delle crisi per almeno due anni
Nelle nostre pazienti è stato riscontrato un uso e un’efficacia maggiore del valproato come monoterapia rispetto a quanto riportato in letteratura.
I farmaci utilizzati per il follow-up sono indicati in tabella 4. In giallo sono cerchiati i farmaci o le associazioni farmacologiche che hanno portato in alcuni casi alla scomparsa delle crisi per almeno due anni, in rosa quelli a seguito dei quali si è registrata una riduzione significativa della frequenza delle crisi. In verde è segnalata un’associazione farmacologica a cui è seguito un aumento delle crisi epilettiche.
Tabella 4

Risposta ai farmaci ed effetti collaterali riscontrati:
| Farmacoresponsive |
18/38 (47%) |
| Farmacoresistenti |
20/38 (53%) |
Non abbiamo dati sufficienti per poter valutare l’efficacia nelle nostre pazienti della dieta chetogena o della stimolazione vagale, che vengono segnalati come possibili approcci terapeutici anche nelle pazienti con SR che presentano un’epilessia farmacoresistente.
Gli effetti collaterali che abbiamo riscontrato sono globalmente modesti:
| Effetti collaterali |
Casi e farmaci |
| Diminuzione appetito |
1 (VPA) |
| Irrequietezza |
4 (PRM, LEV, LTG, TPM) |
| Tremore alle mani |
1 (VPA) |
| Perdita di capelli |
1 (VPA) |
| Anoressia |
1 (TPM) |
Si sottolinea come più farmaci hanno provocato irrequietezza con eccitamento psicomotorio ad attestare la bassa soglia di questo fenomeno nelle pazienti con SR.
Dai dati preliminari ottenuti la presenza di epilessia non sembra essere correlata in modo univoco né al fenotipo clinico (classico-variante) né al tipo di mutazione o dominio funzionale coinvolto. Tuttavia si può rilevare una maggior frequenza di epilessia in soggetti associata ad alcune mutazioni (R294X). I dati non sono ancora sufficienti per correlare mutazioni MeCp2 con la frequenza delle crisi.
Nelle pazienti a linguaggio conservato l’epilessia è presente con frequenza minore rispetto alle forme classiche e spesso è lieve.
Le forme con una compromissione clinica maggiore presentano una frequenza maggiore di epilessia.
Considerazioni conclusive e prospettive future:
L’approfondimento della storia naturale dell’epilessia e della risposta ai farmaci nella Sindrome di Rett ha la finalità di condurre a maggiori conoscenze in questo ambito, soprattutto per il rapporto con il genotipo e con la compromissione clinica, unitamente a promuovere un avanzamento nella presa in carico della singola paziente.
I dati preliminari raccolti da tre centri meritano un ampliamento con l’apporto delle casistiche degli altri centri coinvolti: ciò costituisce l’attività di ricerca che il gruppo si è impegnato ad effettuare.
BIBLIOGRAFIA
- Huppke P, Köhler K, Brockmann K, Stettner GM, Gärtner J. Treatment of epilepsy in Rett syndrome. Eur J Paediatr Neurol. 2007; 11:10-6.
- Jian L, Nagarajan L, de Klerk N, Ravine D, Bower C, Anderson A, Williamson S, Christodoulou J, Leonard H. Predictors of seizure onset in Rett syndrome. J Pediatr. 2006; 149: 542-7.
- Goyal M, O’Riordan MA, Wiznitzer M. Effect of topiramate on seizures and respiratory dysrhythmia in Rett syndrome. J Child Neurol. 2004; 19: 588-91.
- Steffenburg U, Hagberg G, Hagberg B Epilepsy in a representative series of Rett syndrome. Acta Paediatr. 2001; 90: 34-9.
- Nieto-Barrera M, Nieto-Jiménez M, Díaz F, Campaña C, Sánchez ML, Ruiz del Portal L, Siljeström ML.Clinical course of epileptic seizures in Rett’s syndrome Rev Neurol. 1999; 28: 449-53.
- Glaze DG, Schultz RJ, Frost JD. Rett syndrome: characterization of seizures versus non-seizures. Electroencephalogr Clin Neurophysiol. 1998; 106: 79-83.
- Stenbom Y, Tonnby B, Hagberg B.Lamotrigine in Rett syndrome: treatment experience from a pilot study. 13: Eur Child Adolesc Psychiatry. 1998; 7: 49-52.
- Uldall P, Hansen FJ, Tonnby B. Lamotrigine in Rett syndrome. Neuropediatrics. 1993; 24: 339-40.
