
Gli studi genetici pubblicati negli ultimi mesi: le novità, le conferme e gli indirizzi per il futuro della ricerca genetica a livello mondiale.
Silvia Russo, Istituto Auxologico Italiano, Milano
In circa l’80% dei casi, considerando la totalità dello spettro clinico della sindrome di Rett (RTTS), la presenza di mutazioni nel gene MeCP2 (Methyl CpG binding protein 2) è responsabile dell’insorgenza della malattia. Per molto tempo si è pensato che la proteina riconoscesse sequenze metilate a monte di specifici geni, geni target, inibendone l’espressione ed esercitando così un ruolo di repressore trascrizionale. L’identificazione dei geni target è da sempre uno dei principali obiettivi della ricerca in quanto permetterà di capire meglio la correlazione tra la presenza delle mutazione in MeCP2 e le manifestazioni cliniche della RTTS e di pensare anche a come eventualmente sopperire nelle pazienti all’eccesso o deficit di determinate proteine.
Tuttavia nonostante numerosi studi con microarry, che permettono di valutare in contemporanea l’espressione genica di un numero elevatissimo di geni, lo scenario al proposito appariva piuttosto confuso con risultati contradditori nei diversi esperimenti. In una recente review
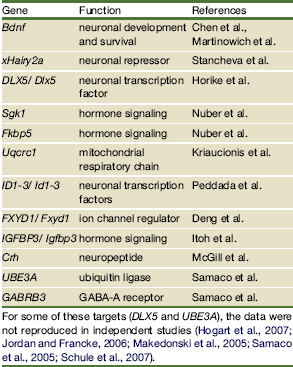
Chahrour e Zoghbi riportavano la tabella qui sopra: alcuni dei targets identificati, ma solo alcuni, sono stati confermati in modo univoco. La difficoltà incontrata nell’identificazione dei target di MeCP2 nasceva dalla non corretta interpretazione del ruolo di MeCP2 e dalla non selettività del tessuto studiato. Infatti il primo mappaggio su larga scala dei siti di legame di MeCP2 nel tessuto neuronale eseguito considerando 26,5 Mb di isole CpG in geni imprinted e non imprinted (Yasui et al 2007) ha rivelato che il 59% dei siti di legame di MeCP2 è situato esternamente ai geni (intergenico) o negli introni e solo il 6% nelle isole CpG. Utilizzando un approccio integrato che tiene conto sull’intero genoma del legame di MeCP2 ai promotori, della metilazione alle isole CpG e dell’espressione genica, si è evidenziato che il 63% dei promotori legati a MeCP2 sono attivamente espressi, mentre il 6% sono altamente metilati. Si è riscontrata una concordanza per questi dati in esperimenti condotti su linee cellulari neuronali umane e su cervello di topo. I nuovi risultati hanno permesso di capire che i targets di MeCP2 non sono solo i geni non espressi, ma anche geni attivi e quindi che la corretta definizione di MeCP2 non è quella di repressore trascrizionale, ma di regolatore trascrizionale.
 La Dottoressa Silvia Russo durante il suo intervento al convegno
La Dottoressa Silvia Russo durante il suo intervento al convegno
E’ in tale contesto che si inserisce lo studio, apparso sulla rivista Science a fine maggio 2008, eseguito nel laboratorio della dott.ssa Zoghbi. Punto di partenza dello studio è stata la considerazione che sia l’assenza di MeCP2, sia la duplicazione con overespressione dello stesso gene determinavano l’insorgenza di un quadro neurologico gravemente compromesso, seppure diverso, e la disponibilità sia di un modello murino che non produce Mep2, Mecp2-null, sia di un modello murino che overesprime MeCP2 sotto il controllo di un promoter endogeno (Mecp2-Tg). Precedenti analisi dei profili trascrizionali del materiale genetico estratto dai cervelli dei due modelli avevano evidenziato piccole differenze nell’espressione genica ed è perciò che Chahrour e colleghi hanno pensato di concentrarsi sullo studio di una precisa regione del cervello, l’ipotalamo, che sembra correlata specificamente a molti sintomi che si osservano nella sindrome di Rett (ansietà, decelerazione dell’accrescimento, disturbi del ritmo sonno-veglia, e alterazioni del sistema nervoso autonomico). Sia nel topo MeCP2-null, sia nel topo MeCP2-Tg si osservavano migliaia di geni disregolati, ma inaspettatamente l’85% (2184/2582) erano attivati da MeCP2, mentre solo 377 erano repressi, risultato in perfetto accordo con la definizione di regolatore trascrizionale.
Inoltre l’osservazione di 1187 geni con espressione alterata solo in MeCP2-Tg e 369 con MeCP2 alterato nel topo null fa supporre l’esistenza di target secondari i cui cambiamenti dipendono dalle variazioni nei target primari.
Inoltre l’analisi ontologica dei due gruppi di geni, attivati e repressi, ha evidenziato la loro appartenenza, in termini di funzioni biologiche, a due categorie distinte. Le maggiori variazioni di espressione sono state riportate dai geni che codificano per recettori appaiati alle proteine G, in accordo con il fatto che la maggior parte dei recettori per i neuropeptidi ipotalamici sono GPCRs che appartengono alla lista dei geni attivati da MeCP2; alla lista dei geni repressi appartengono i recettori olfattori. La scoperta che la maggior parte dei geni controllati da MeCP2 nell’ipotalamo sono attivati ha portato all’ipotesi che MeCP2 fosse associato in vivo a coattivatori della trascrizione. Esperimenti di immunoprecipitazione e seqChIP hanno evidenziato l’associazione di MeCP2 e CREB1, sui promotori di geni che vengono attivati da MeCP2 ma su quelli che sono repressi; inoltre si è visto che lo stesso CREB1 è a sua volta un target di MeCP2 che ne promuove l’espressione.
Lo studio della dott.ssa Zoghby è molto importante sia perché dimostra che il ruolo di MeCP2 in uno dei tessuti più importanti per l’espressione clinica della sindrome di Rett è prevalentemente quello di attivatore di espressione e tra i molti geni disregolati, inizia ad indicare alcuni target con funzione correlata, quali A2bp1, che produce una proteina che controlla lo splicing di geni neuronali e la cui assenza è stata riscontrata in soggetti con ritardo mentale ed epilessia, oppure Gamt (guanidinoacetato metiltransferasi), un target attivato da MeCP2 e coinvolto nella biosintesi della creatina, e lo stesso BDNF che viene quindi attivato da MeCP2.
Si apre con queste nuove scoperte una nuova prospettiva per la conscenza dei meccanismi d’azione di MeCP2 e per la ricerca di una soluzione delle principali problematiche della malattia.
Un filone parallelo della ricerca si occupa di stabilire l’esistenza di una relazione tra il quadro clinico della malattia e la presenza di una specifica mutazione di MeCP2. A tutt’oggi sono state descritte oltre 200 diverse mutazioni di MeCP2, ma 8 sono quelle ricorrenti presenti in circa il 70% delle bambine mentre un altro 10% presenta piccole delezioni nella regione C-terminale. Lo studio della correlazione genotipo-fenotipo, che sia di aiuto nello stabilire una prognosi più o meno severa della malattia, necessita di grandi casistiche che permettano di disporre anche per le singole mutazioni di un numero di pazienti statisticamente significativo; inoltre è necessario scegliere una scala di valutazione dei segni clinici il più possibile appropriata.
Lo studio di Babbington, apparso su Neurology in aprile, utilizza 3 diverse scale di severità, Percy, Kerr e Pineda e si avvale della casistica che afferisce al database internazionale InterRett. Secondo questo lavoro indipendentemente dalla scala di valutazione utilizzata, le mutazioni p.R294X e p.R133C risultano essere globalmente non gravi; in altri casi si osserva una discordanza che dipende proprio dai parametri considerati da una data scala di valutazione, ad es le mutazioni pR270X e p.R255X sono le più severe secondo la scala Pineda, (che considera prevalentemente l’evoluzione della malattia), ma se si utilizza la scala di Kerr (che considera lo stato clinico nel momento corrente) la mutazione più severa è considerata la p.T158M.
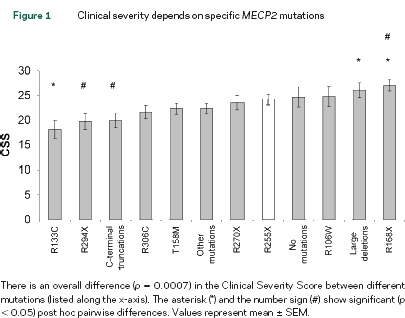
Sempre sulla stessa rivista, Neul ha pubblicato uno studio analogo utilizzando una scala specifica CSS (Clinical Severity Score) per la Rett su un campione di 236 pazienti in cui riscontrava un’effettiva differenza nella severità globale della malattia correlata alla specifica mutazione. I risultati sono riportati nella tabella qui sotto.
Un altro importante strumento della ricerca è la creazione di modelli murini che permettano di affrontare problemi specifici.
Lo scorso anno si è molto parlato dell’esperimento in cui il gruppo del dr Bird (2007) ha creato un topo MeCP2 lox-stop che, dopo aver sviluppato tutti i sintomi della malattia, riattivando il gene riusciva a recuperare una funzionalità normale. Lo studio suggeriva per MeCP2 un ruolo essenziale nel neurone adulto.
Successivamente sono stati creati altri modelli che supportassero questo dato.
Il modello murino descritto da Schmidt, dimostra che MeCP2 è presente in grande quantità nei neuroni maturi; il modello di Samaco, invece, creatosi per la mancata excisione della cassetta PGK-neo nel 3’UTR possiede una minor efficienza di trascrizione del gene e presenta un fenotipo caratterizzato da deficit motori, problemi respiratori, deficit nel comportamento, dimostrando che anche modeste riduzioni della proteina possono comportare problemi del neurosviluppo.
Infine Palmer e colleghi hanno creato un modello murino con una mutazione tardiva MeCP2308/y che ha il vantaggio di sopravvivere a lungo permettendo di studiare fase acuta e cronica della malattia. Interessante anche l’utilizzo del tessuto olfattorio che ha il vantaggio di essere sottoposto ad una continua neurogenesi funzionale: si dimostrano due distinte fasi: a) una fase acuta con un transitorio ritardo nella differenziazione durante sinaptogenesi e b) una fase cronica in cui si verifica la distruzione della funzione che viene compensata dall’incremento dei neuroni immaturi.

Il team della professoressa Lidia Larizza: Serena Ferraiuolo, Ester Mainini, Francesca Cogliati, Margherita Marchi, Maura Masciardri, Renato Lupi, Valentina Giorgini, Miriam Cinque, Silvia Russo
Referenze
- Chahrour M and Zoghbi HY (2007). The Story of Rett Syndrome:From Clinic to Neurobiology. Neuron 56, 422-437
- Palmer A (2007) MeCP2 mutation causes distinguishable phases of acute and chronic defects in synaptogenesis and maintenance, respectively. Mol. Cell. Neurosci. 37 (2008) 794–807
- Yasui DH et al (2007) Integrated epigenomic analyses of neuronal MECP2 reveal a role for long-range interaction with active genes. PNAS 104, 49: 19416–19421
- Chahrour M et al (2008) MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320:1224-1229
- Bebbington A et al(2008) Investigating genotype–phenotype relationships in Rett syndrome using an international data set. Neurology,70:868–875
- Neul J. et al (2008) Specific mutations in Methyl-CpGBinding Protein 2 confer different severity in Rett syndrome. Neurology 70:1313–1321
- Schmid RS et al (2008) A methyl-CpG-binding protein 2-enhanced green fluorescent protein reporter mouse model provides a new tool for studying the neuronal basis of Rett syndrome. Neuroreport. 5;19(4):393-8
- Samaco RC et al (2008)A partial loss of function allele of methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndromeHum Mol Genet. 15;17(12):1718-27
