La relazione esposta durante il convegno di Cervia si poneva gli obiettivi di riferire i risultati del progetto finanziato
da AIRett nel precedente bando di ricerca 2017-2019, il cui scopo era generare un modello in vitro di neuroni corticali derivati da bambine portatrici delle mutazioni più frequenti come causa della Sindrome di Rett e di illustrare come i dati ed i modelli biologici, frutto del primo progetto, possano essere un punto di partenza per valutare le potenzialità di farmaci capaci di riattivare l’allele sano di MeCP2 sul cromosoma X. È importante sottolineare come sia la generazione, sia l’identificazione di modelli in vitro, sia la riattivazione del cromosoma si inseriscono nella mappa tracciata per la ricerca sulla Sindrome di Rett a livello internazionale. Sebbene neuroni derivati da cloni di IPSC umani siano stati già generati in precedenza da altri gruppi di ricerca, non sono riportate molte delle mutazioni più ricorrenti tra le bimbe Rett, quindi ci siamo proposti di produrre un modello di mutazioni ricorrenti scegliendole in modo che fossero rappresentative anche delle diverse manifestazioni cliniche della malattia: p.Arg168ter (R168X) considerata tra le più severe, p.Arg255ter (R255X) moderata e p.Arg133Cys (p.R133C) che spesso permette un miglior decorso clinico. L’esperimento richiede un iniziale prelievo di sangue, da cui vengono separati solo i globuli bianchi che successivamente verranno indotti, mediante trasfezione con un vettore di specifici geni a trasformarsi in cellule staminali pluripotenti, IPSC. Le cellule così ottenute sono cellule multipotenti proprio come le cellule staminali e possono quindi essere indotte a differenziarsi in quasi qualunque tipo cellulare, nel nostro caso in neuroni della corteccia frontale. Poiché le mutazioni oggetto dello studio sono sul cromosoma X e poiché tale cromosoma è soggetto nelle donne ad un meccanismo di “inattivazione selettiva del cromosoma X”, in ciascuna cellula solo uno dei cromosomi X è attivo e produce la proteina. I cloni ottenuti da ciascuna bimba dovrebbero esprimere o solo l’allele mutato oppure solo l’allele sano e questi ultimi sono un ottimo controllo sano, in quanto differiscono dagli altri solo per il difetto nel gene MeCP2. Si parla di controlli “isogenici”.
L’idea era ottenere cloni IPSC con e senza il difetto genetico in neuroni e valutarne le differenze morfologiche e funzionali. Ci si proponeva di studiare tre pazienti per ciascuna mutazione. Abbiamo raggiunto l’obiettivo per la variante R133C associata al quadro clinico più lieve, mentre studiando pazienti con le altre 2 mutazioni, abbiamo verificato che in più casi nel sangue si esprimeva solo l’allele sano, quindi non si potevano ottenere neuroni “malati”. Questo riscontro ci fa ipotizzare che in alcune pazienti sia presente un mosaicismo di tessuto che forse potrebbe avere un ruolo protettivo nelle mutazioni più gravi. Grazie alla collaborazione con le famiglie di AIRett, studiando 5 pazienti siamo riusciti ad ottenere 2 casi con R255X, che esprimono la mutazione nel sangue ed in se- guito alla diagnosi di un maschio con variante p.Gly252ArgfsTer7 (in una posizione quindi molto vi- cina all’aminoacido 255), siamo riusciti a raggiungere un numero di pazienti – ovvero 3 – necessa- rio perchè i nostri risultati possa- no essere significativi. Più difficile la situazione per il difetto R168X, perché tutte le bambine che ab- biamo prelevato presentavano un’inattivazione selettiva del cro- mosoma X. Solo in un caso la ri- programmazione, pur partendo da cellule sane, ha prodotto clo- ni che esprimevano solo l’allele mutato, modificazione che si è conservata durante i passaggi di differenziamento in neuroni. A tal proposito, sebbene il progetto sia ufficialmente chiuso, se qualche famiglia volesse aiutarci vorrem- mo riuscire a generare anche il modello della R168X.
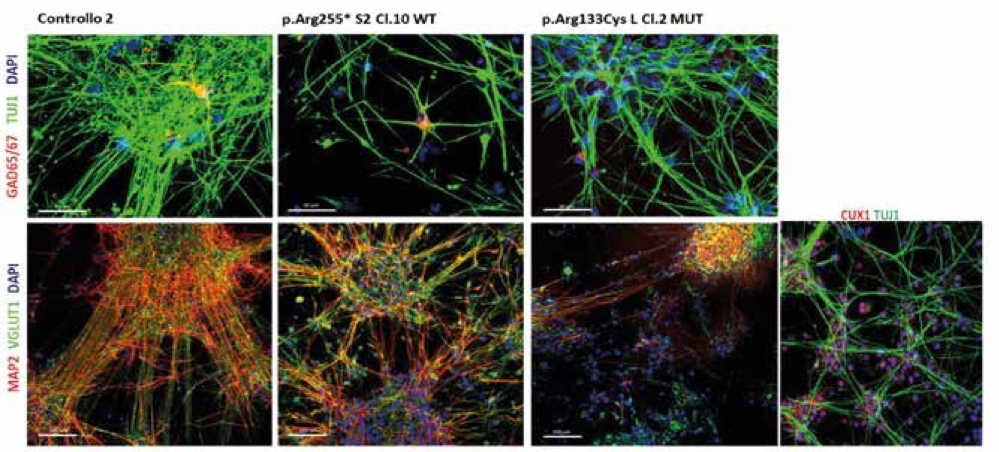
Dopo aver controllato nei cloni di IPSC la staminalità (valutando che le cellule fossero effettivamente in grado di differenziarsi nei tre foglietti embrionali e di esprimere i marcatori della totipotenza) e la stabilità genomica (per verificare che nel corso della complessa sperimentazione non fossero insorte delle alterazioni a livello genomico che potessero modificare il comportamento delle cellule), i neuroni sono stati differenziati in neuroni corticali e verificata la presenza di marcatori specifici per la componente eccitatoria (VGLUT1) ed inibitoria (GAD65/67) dei markers presinaptici (SYP) dei markers tipici della corteccia CUX1 e TUJ1 delle pazienti Rett sono stati tutti caratterizzati dimostrando che la loro espressione sia nei neuroni sani che quelli che portavano il difetto genetico (Fig. 1).
Venendo quindi allo scopo principale del lavoro abbiamo valutato le differenze morfologiche, elettrofisiologiche e biochimiche tra i neuroni con la mutazione in MeCP2 ed i loro controlli isogenici. Le valutazioni morfologiche hanno evidenziato una ridotta dimensione nel nucleo dei neuroni mutati. L’analisi eseguita a due diversi tempi del differenziamento, precisamente a 42 giorni e a 70 giorni, mostra come le significatività siano più consistenti con lo sviluppo dei neuroni e come si possano osservare più tardivamente nei neuroni con la mutazione meno severa, R133C (Fig. 2).
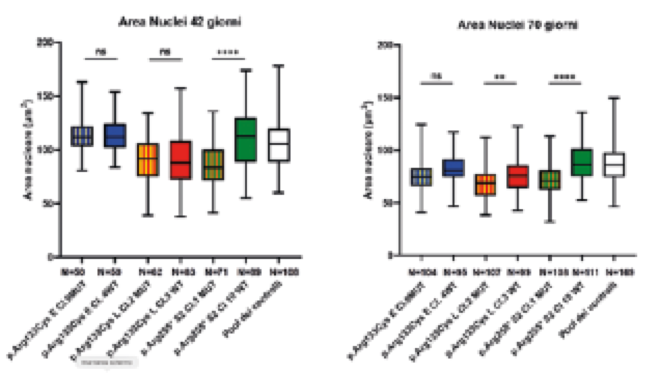
I grafici rappresentano le aree dei nuclei delle cellule neuronali, rispettivamente in uno stadio precoce del differenziamento (42 giorni) ed in uno stadio più avanzato (70 giorni). Ogni mutata accanto al proprio controllo, N è il numero di cellule misurate. Gli asterischi in alto indicano una differenza significativa tra il neurone con la mutazione e quello sano.
Si vede come le differenze siano più evidenti nella mutazione p.Arg255* e come la significatività per p.Arg133Cys si cominci ad osservare solo ad un tempo di maturazione più avanzato. Non sono state osservate differenze significative invece nella lunghezza e ramificazione dei neuroni a 42 giorni, unico timepoint in cui sia-
mo riusciti ad eseguire le analisi, perché con il procedere del differenziamento la rete diviene intricata e non è possibile distinguere i singoli neuroni.
In collaborazione con il prof. Pizzorusso del CNR di Pisa ed il suo dottorando dott. Gianluca Pietra sono state indagate le proprietà elettrofisiologiche mediante registrazione del potenziale di membrana e delle correnti ioniche dei neuroni maturi a 110-120 giorni,comparando per ciascuna paziente il comportamento dei neuroni sani, che non esprimono la mutazione, ed i neuroni che la esprimono. Il metodo utilizzato è il patchclamp, che va a registrare l’attività delle singole cellule neuronali. Questi dati sono molto importanti perché misurando l’attività elettrica dei neuroni ne valutano la funzionalità, la capacità di trasmettere segnali e di comunicare con gli altri neuroni. I parametri studiati sono: capacità di membrana, che fornisce anche una misura indiretta della superficie della cellula e quindi delle sue arborizzazioni, resistenza di membrana, costante di tempo, potenziale di membrana, corrente sodio voltaggio dipendente, corrente potassio voltaggio dipendente e la frequenza di correnti sinaptiche spontanee, sia inibitorie che eccitatorie. Ad oggi sono stati studiati i neuroni derivati dai cloni isogenici di 2 pazienti con la mutazione p.Arg133Cys, e di una paziente con la mutazione p.Arg155*, mutazioni come abbiamo detto con un diverso comportamento dal punto di vista clinico. Una prima importante osservazione, che ci ha reso confidenti dei nostri dati seppure ancora preliminari, è il fatto che neuroni derivati dai cloni sani di ciascuna paziente producono dati sovrapponibili, mentre le due mutazioni sembrano comportarsi in modo diverso.
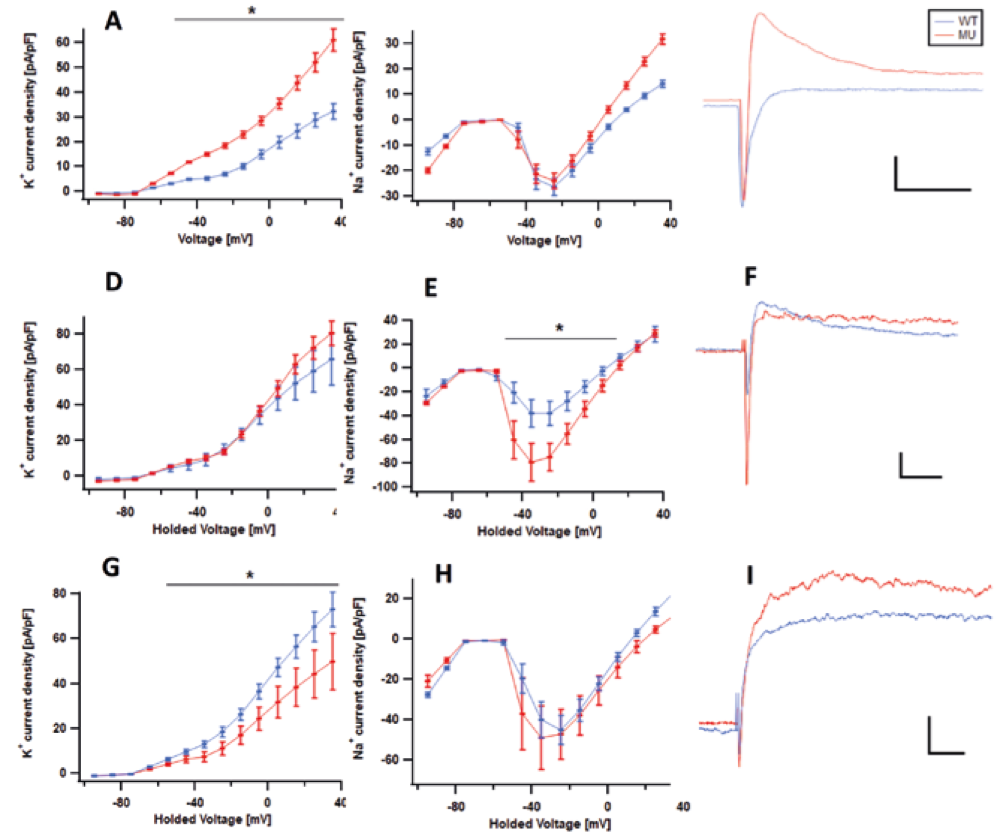
Nei neuroni di controllo rispetto a quelli che esprimono la mutazione p.Arg155* si osserva coerentemente una differenza di maturazione neuronale; le proprietà passive della membrana tra cui la capacità, la resistenza ed il potenziale di membrana mettono in evidenza una minore maturazione delle cellule mutate rispetto alle WT. Nelle due pazienti con la mutazione p.Arg133Cys, si osserva invece un’accelerazione dello sviluppo ma con ridotta espressione dei canali Na+ voltaggio dipendenti.
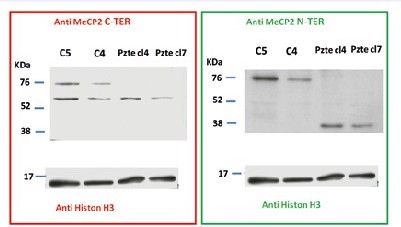
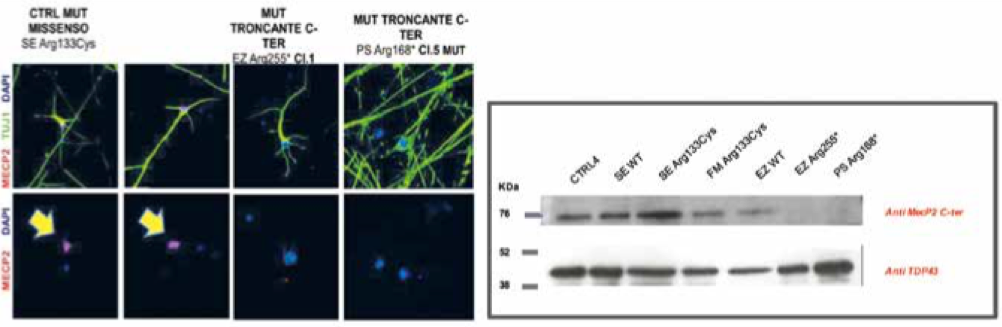
Al fine di verificare l’assenza della proteina nei neuroni con l’allele difettivo per la proteina MeCP2 e la presenza nei cloni con l’allele sano sono stati eseguiti esperimenti di western blot e di immunofluorescenza che riconoscono la porzione terminale della proteina. Nei neuroni della paziente con mutazione p.Arg255* il segnale è assente perché quella porzione della proteina è mancante a causa del difetto genetico, mentre nei neuroni di controllo è visibile e si può verificare che localizza, come atteso in modo specifico nel nucleo. Questo esperimento è molto importante perché ci permetterà di verificare nel nostro modello se un farmaco sarà in grado di riattivare l’allele sano, nelle cellule che esprimono l’allele con la mutazione. Nel caso della variante p.Arg133Cys, poiché la proteina non è mancante in nessuna sua parte ma diversa in un solo aminoacido, non si osservano differenze con questo metodo.
Inoltre per verificare se la mutazione determini la produzione di una proteina tronca oppure la proteina venga degradata, abbiamo ripetuto questo esperimento con un anticorpo che riconosca la porzione N-terminale (iniziale) della proteina MeCP2 nei neuroni derivati dal paziente di sesso maschile. In questo caso mentre con l’anticorpo C-terminale non si vede nulla, come nella paziente p.Arg255*, nell’anticorpo N-terminale si osserva una banda più in basso, quindi più corta, a riprova che questa proteina non viene degradata. C5 e C4 sono neuroni derivati con lo stesso identico protocollo da individui sani di controllo. Dovremo ora verificare in immunofluorescenza se tale proteina tronca entri o meno nel nucleo.
A livello di cellule staminali, primo passaggio verso la generazione di neuroni delle pazienti, la quantità di proteina MeCP2 che viene prodotta in ciascun allele è molto ridotta e non si potranno usare queste metodiche per verificarne la riattivazione. La riattivazione del cromosoma X verrà verificata nell’RNA dei diversi cloni dove dovremo verificare la comparsa anche in piccole quantità del trascritto sano accanto a quello mutato.