
Enrico Tongiorgi
Dipartimento di Scienze della Vita, Università di Trieste
Parte 1. Strategie terapeutiche e sperimentazioni cliniche nella sindrome di Rett: il punto della situazione.
La sindrome di Rett presenta delle notevoli sfide per la ricerca di una sua possibile cura a causa delle sue mille sfaccettature e dei tanti diversi sintomi, così variabili tra una bambina e l’altra. Molte speranze si sono accese negli ultimi anni a seguito alla scoperta che nei topi MeCP2y/-(Null), modello della sindrome di Rett “classica”, la reintegrazione del gene MeCP2 normale – in grado di contrastare i principali sintomi della malattia (Guy et al. 2007). Per questo, nonostante le oggettive difficoltà, molti ricercatori sono convinti che si possa trovare una “chiave” capace di innescare la via molecolare per la riattivazione del normale programma di sviluppo che si era bloccato per l’assenza del gene MeCP2. Ma qual è la “chiave” giusta per risvegliare il programma di crescita interrotto dalla sindrome di Rett?
Ispirati da questa domanda scientifica, i ricercatori hanno definito delle strategie per lo sviluppo di una terapia seguendo sostanzialmente due strade principali, una “a monte”, l’altra “a valle” del gene MeCP2 (Katz et al., 2016). Nella prima strategia (a monte), si cerca di ripristinare il normale funzionamento del gene MeCP2, nella seconda (a valle) si mira a potenziare particolari vie metaboliche che vengono attivate a cascata dal gene MeCP2 (Figura 1).
Più in dettaglio, quando il bersaglio della terapia è il ripristino del MeCP2, sono state intraprese 4 diverse strategie:
1) L’attivazione del gene MeCP2 sano posto su uno dei due cromosomi X che è “silente”. Tutte le bambine hanno due cromosomi X (femminili), ma ogni cellula dell’organismo ne usa solo uno e quindi l’altro cromosoma X viene “silenziato”, dividendo circa al 50% l’uso di ciascun cromosoma X. Nella sindrome di Rett un cromosoma X porta il gene mutato, l’altro, il gene sano e quindi in modo del tutto casuale, alcune cellule del corpo sono sane e altre sono malate. Una terapia ideale dovrebbe agire soltanto sulle cellule malate, riattivando il gene sano silenziato.
2) L’espressione del gene MeCP2 sano mediante vettori di terapia genica. Si tratta in genere di trasferire il gene mediante un piccolo virus, innocuo, che penetra nelle cellule dell’organismo e produce i propri geni tra i quali si può inserire artificialmente il gene di interesse o un gene che è in grado di riparare il gene mutato (tecniche di genome editing, o di mRNA editing).
3) La somministrazione della proteina mancante. Questa terapia, è stata sperimentata con successo nei topi privi del gene CDKL5 a cui è stata iniettata la proteina umana CDKL5. Questa proteina-surrogato, grazie ad una appendice costituita da una piccola catena di aminoacidi, viene trasportata naturalmente all’interno delle cellule mantenendone la funzione (Brevetto US20150247134 A1 depositato da Elisabetta Ciani e Franco Laccone, Università di Bologna).
4) Composti che permettono ai ribosomi di produrre la proteina “saltando” le mutazioni di stop (read-through compounds). In molte pazienti Rett la mutazione di una singola lettera del codice genetico genera un segnale di STOP prematuro, cioè posizionato nel cuore della sequenza del gene MeCP2 anziché al suo termine, come avviene normalmente. Perciò, quando il DNA che porta la sequenza con la mutazione di STOP viene letto dai ribosomi per produrre la proteina corrispondente, si ottiene una catena proteica più corta e generalmente priva delle funzioni di MeCP2 utili alla cellula. Alcuni composti sperimentati recentemente, permettono ai ribosomi di leggere la sequenza del DNA in modo meno preciso e quindi di inserire un aminoacido “utile” anche quando trovano un segnale di STOP prematuro, rispettando tuttavia il segnale di STOP canonico presente alla fine di una sequenza del codice da cui viene prodotta la proteina.
Ciascuna di queste strategie porta con sé delle problematiche tecniche nell’applicazione alla sindrome di Rett, dove solo la metà delle cellule del corpo sono colpite dalla malattia. Inoltre la quantità di proteina MeCP2 presente nelle cellule deve essere contenuta entro dei limiti ben precisi. Infatti, non solo bassi livelli o la completa assenza della proteina MeCP2 sono patologici per l’organismo, causando la sindrome di Rett, ma anche quantità in eccesso provocano una sindrome conosciuta come MeCP2-duplication syndrome.
Quindi la terapia dovrebbe essere in grado di ripristinare in modo selettivo il gene soltanto nelle cellule in cui – presente il gene mutato ed il gene sano è inattivato. Infatti, se usando la terapia genica (strategia 2) venisse inserito un gene soprannumerario nelle cellule “sane” si potrebbe generare la sindrome di duplicazione. Un problema simile si potrebbe creare anche nel caso della somministrazione di proteina surrogata (strategia 3). Dato che non è mai stata testata nella Rett “classica” gli effetti di una terapia proteica con MeCP2 non sono noti, mentre nel caso dell’CDKL5 sembra produrre degli effetti benefici nel modello animale della sindrome. Analogamente, riattivando l’intero cromosoma X (strategia 1), si potrebbe generare un effetto di duplicazione per tutti gli altri geni sani del cromosoma X. Nel caso invece, in cui la terapia genica venisse utilizzata per riparare il gene mutato, ci si attende un risultato più mirato ed una minore tossicità, sebbene le tecnologie disponibili (per esempio la tecnica CRISPR/CAS9) abbiano dimostrato finora una limitata efficacia. Per quanto riguarda i farmaci che permettono ai ribosomi di saltare le mutazioni di STOP (strategia 4), si tratta di un’idea che esiste da alcuni decenni e recentemente è stato prodotto un gran numero di nuovi composti chimici destando un notevole interesse in quanto alcuni sono risultati efficaci nel ripristinare almeno parzialmente le funzioni di vari geni mutati. Alcune sperimentazioni cliniche sono già in stadio avanzato anche per malattie legate a geni del cromosoma X, anche se bisogna ammettere che esistono solo pochi composti veramente efficaci e pertanto questa terapia ha ancora un uso clinico limitato. Infatti, la capacità di un composto di saltare la mutazione STOP dipende molto dal tipo di mutazione. Questi composti risultano efficaci solo per alcuni pazienti mentre per altri pazienti, con altre mutazioni, non risultano utili. Pertanto c’è la necessità di disegnare nuovi composti “read-through” affinché siano attivi su tutte le varie tipologie di mutazioni. Le ricerche attuali si incentrano anche sul risolvere il problema dell’inserimento di un aminoacido sbagliato che potrebbe causare una disfunzione della proteina, o potrebbe produrre una proteina instabile.
La seconda strada per lo sviluppo di nuove terapie, prevede l’uso di farmaci che attivano le vie metaboliche a valle del gene MeCP2 e consiste in 4 strategie principali (Figura 1):
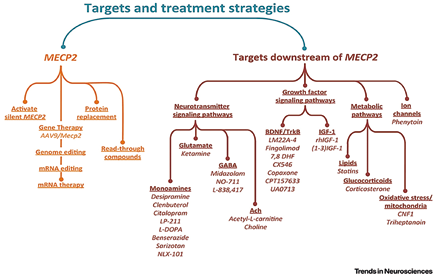
Figura 1. Da: D.Katz, A.Bird, M.Coenraads, S.J. Gray, D.U. Menon, B.D. Philpot, and D.C. Tarquinio (2016). Rett Syndrome: Crossing the Threshold to Clinical Translation. Trends Neurosci. 39(2): 100–113.
1) Farmaci che modulano i neurotrasmettitori e le loro vie di segnale. Sono stati sperimentati trattamenti per aumentare i livelli di Acetilcolina, Noradrenalina, Serotonina, Dopammina, Glutammato e Acido-Gamma-Ammino-Butirrico (GABA). Per le monoammine (Noradrenalina, Serotonina) sono stati utilizzati antidepressivi, mentre per la Dopammina, la levo-Dopa da sola o in combinazione con un inibitore della dopa-decarbossilasi (Szczesna et al., 2014). Per aumentare la produzione dell’Acetilcolina è stata somministrata la Colina che è il nutriente da cui viene sintetizzato questo neurotrasmettitore (Nag e Berger-Sweeney, 2007). Per il Glutammato è in corso di sperimentazione clinica la Ketamina (vedi di seguito).
2) Farmaci che surrogano o potenziano i fattori neurotrofici. I fattori neurotrofici bersaglio di questa strategia terapeutica sono sostanzialmente due: la neurotrofina Brain-Derived Neurotrophic Factor (BDNF) e l’Insulin-like Growth Factor-1 (IGF-1). Entrambi sono in grado di promuovere la crescita dei neuroni Rett mediante il ripristino di vie di segnale in parte sovrapponibili, ed entrambi promuovono la riduzione di vari sintomi, incluso le apnee respiratorie. Tuttavia mentre la somministrazione di BDNF è poco efficace perché non raggiunge il cervello in quantità sufficienti e quindi è necessario usare dei famaci che attivano il suo recettore, l’IGF-1 raggiunge bene il sistema nervoso ed è oggetto di sperimentazione clinica (vedi di seguito).
3) Farmaci che modulano i lipidi, gli steroidi endogeni o proteggono dallo stress ossidativo. In questa categoria rientrano diverse strategie che hanno un obiettivo comune: una migliore produzione di energia attraverso una efficiente metabolizzazione degli acidi grassi, riducendo al contempo lo stress ossidativo che è normalmente associato alla produzione di energia e che nella sindrome di Rett risulta superiore ai livelli normali (Leoncini et al., 2015). Si ritiene che tale strategia possa avere degli effetti benefici per il trattamento delle crisi epilettiche.
4) Farmaci che ripristinano il funzionamento dei canali ionici. Precedenti approcci sono stati mirati ai canali coinvolti nell’epilessia, come il canale per il flusso degli ioni Sodio. Recentemente si è scoperto che un canale al Potassio/Cloro, noto con la sigla KCC2, è quasi assente nei neuroni Rett causando una ipereccitazione dei neuroni che sarebbe alla base delle crisi epilettiche (Tang et al., 2016). È stato inoltre dimostrato che il ripristino dei livelli normali di KCC2 permette ai neuroni Rett di maturare verso un fenotipo adulto, bilanciando correttamente segnali di tipo inibitorio (mediati dal neurotrasmettitore GABA) e quelli di tipo eccitatorio (mediati dal Glutammato) (Tang et al., 2016). Con livelli normali di KCC2, i neuroni sarebbero più efficaci nel trasmettere i segnali (Glutammato) mantenendo tuttavia un corretto freno inibitorio (GABA) che evita ai neuroni di eccedere verso la generazione di correnti elettriche incontrollate di tipo epilettico.
Le sperimentazioni cliniche con la Colina, per aumentare i livelli di Acetilcolina o con Levo-Dopa per aumentare la produzione di Dopammina hanno prodotto risultati modesti (strategia 1). Mentre gli antidepressivi sono ancora in fase di sperimentazione e c’è una maggiore aspettativa di ottenere dei successi (strategia 1). Vi è un certo ottimismo anche per quanto riguarda i trattamenti per aumentare i segnali indotti dai fattori neurotrofici (strategia 2) mentre per i trattamenti con integratori alimentari (per es. Omega 3 e 6) e farmaci che regolano il metabolismo energetico dei mitocondri è necessario attendere l’esito delle sperimentazioni in corso (strategia 3). Infine per quanto riguarda i canali ionici (strategia 4), non esiste ancora una sperimentazione clinica mirata al ripristino del canale del Potassio/Cloro KCC2, e il candidato più promettente è la Fenitoina, un farmaco già presente sul mercato ed usato come antiepilettico che ha come target il canale per gli ioni Sodio.
Da notare che a parte alcuni composti nuovi, nella maggior parte dei casi sopra esposti si tratta di farmaci già in commercio e somministrati per altre patologie, i quali tuttavia hanno dimostrato un effetto benefico nei modelli animali della sindrome di Rett. L’uso di “vecchi” farmaci per nuove applicazioni terapeutiche si definisce “strategia di riposizionamento” del farmaco. Ha il vantaggio di permettere una più rapida approvazione da parte delle autorità preposte al controllo dell’uso e al commercio dei farmaci omettendo la Fase 1 della sperimentazione clinica dedicata ai controlli di sicurezza, in ragione del fatto che i principali dati del farmaco sono già noti, come ad esempio quelli del metabolismo del composto attraverso i vari distretti del corpo (cervello, cuore, stomaco, fegato, reni, etc…). Tuttavia, dopo aver dimostrato effetti positivi nel modello animale, restano da dimostrare l’efficacia, l’entità di eventuali effetti collaterali e la tollerabilità specifica nelle pazienti affette dalla sindrome di Rett, soprattutto se in età pediatrica. Nella prima sperimentazione clinica per un farmaco “riposizionato” si può quindi saltare la Fase 1, e viene di norma condotta una Fase 2 dove per precauzione vengono coinvolti solo pochi pazienti in un numero generalmente compreso tra 10 e 40, o un numero doppio se alcuni pazienti ricevono il placebo. Nelle malattie rare molto spesso, la Fase 2 viene condotta dichiarando apertamente quali sono i pazienti che ricevono il farmaco (sperimentazione “Open label“). Solo dopo una prima valutazione del farmaco, se si dimostra una buona tollerabilità e c’è stata anche una minima efficacia si procede ad una sperimentazione su un numero maggiore di pazienti (circa un centinaio) in una cosiddetta Fase 3. Nelle tabelle che seguono vengono schematicamente riportati i principali “clinical trials” con indicazione della fase in cui si trovano (fonte: sito web NIH clinical trials).
Parte 2. Il nostro approccio: Mirtazapina
L’osservazione di una riduzione dei neurotrasmettitori noradrenalina (NE), serotonina (5HT) e dopamina (DA), ha suggerito alcuni anni fa la possibilità di utilizzare gli antidepressivi nella sindrome di Rett (Zoghbi et al., 1985, Ide et al., 2005; Santos et al., 2010). Nel Laboratorio di Neurobiologia Cellulare e dello Sviluppo dell’Università di Trieste abbiamo intrapreso uno studio di riposizionamento selezionando Mirtazapina, un farmaco antidepressivo che potenzia il rilascio dei neurotrasmettitori noradrenalina (NE), serotonina (5HT) e in misura minore anche dopamina (DA).
Il primo antidepressivo testato nella sindrome di Rett è stato la desipramina, un antidepressivo triciclico che inibisce la ricaptazione di NE e in minor misura di 5HT. Nei topi, la desipramina stabilizza la respirazione, sostenendo la crescita dei neuroni dei centri del respiro, ed aumenta l’aspettativa di vita. Sebbene la desipramina non determini un recupero di peso del cervello, del corpo e dell’attività motoria, questi esperimenti hanno chiaramente indicato che un trattamento farmacologico mirato ai sistemi serotoninergico e noradrenergico potrebbe essere d’aiuto per le pazienti Rett (Roux et al., 2007, Zanella et al., 2008). Purtropppo la desipramina ha gravi effetti collaterali, che comprendono l’incapacità di controllare la pressione sanguigna e il battito cardiaco, con improvviso arresto cardiaco in bambini e adolescenti con prolungati intervalli QT, una caratteristica tipica della sindrome di Rett (Pacher and Kecskemeti, 2004). In alternativa alla desipramina, abbiamo scelto un farmaco approvato dalla Food and Drug Administration (FDA) statunitense e dall’European Medicine Agency (EMA): Mirtazapina, un antidepressivo noradrenergico e serotoninergico specifico tetraciclico (NaSSA) che presenta la più alta tollerabilità se paragonato ad altri 12 principali antidepressivi (Cipriani et al., 2009). Mirtazapina è virtualmente priva di effetti collaterali di tipo anticolinergico (Burrows and Kremer, 1997) e ha pochi effetti collaterali cardiorespiratori, sebbene un sovradosaggio induca sedazione (Hartmann, 1999).
I nostri esperimenti, pubblicati recentemente (Bittolo et al., 2016), hanno dimostrato che in topi maschi MeCP2 knock-out (MeCP2y/-), 2 settimane di trattamento con Mirtazapina (dal giorno postnatale P28 al giorno P42) sono sufficienti a recuperare sia i deficit della frequenza cardiaca che quella respiratoria, senza effetti collaterali sulla saturazione d’ossigeno del sangue arterioso né sulla distensione venosa (Bittolo et al., 2016). Mirtazapina può quindi recuperare il ritmo irregolare della respirazione descritto sia nelle pazienti Rett sia nei topi senza provocare gli effetti collaterali indesiderati a livello cardiovascolare osservati con la desipramina (Bittolo et al., 2016). È importante sottolineare come i nostri esperimenti abbiano anche dimostrato che la Mirtazapina è in grado di recuperare l’atrofia cerebrale, come indicato dalle misurazioni del peso del cervello e dalle analisi quantitative della morfologia della corteccia somato-sensoriale (Bittolo et al., 2016). Le misure della morfologia corticale rappresentano un parametro molto robusto per valutare l’efficacia del trattamento farmacologico, in quanto le alterazioni della morfologia corticale nei topi maschi MeCP2y/- e nelle femmine MeCP2+/- sono altamente riproducibili in tutti i laboratori, incluso il nostro, e rappresentano molto verosimilmente le alterazioni della morfologia corticale osservate nelle pazienti (Katz et al., 2012). In particolare, abbiamo riscontrato che il trattamento con Mirtazapina ripristina lo spessore della corteccia somato-sensoriale, che era ridotto del 20% nei topi MeCP2y/-, e ristabiliscono la citoarchitettura degli strati corticali. Questi effetti macroscopici della Mirtazapina sono associati ad un completo recupero della normale struttura microscopica dei neuroni piramidali degli strati II e III. In particolare abbiamo riscontrato un recupero della grandezza del soma, del diametro dei dendriti apicali, dell’arborizzazione dei dendriti basali e della densità delle spine, che risultavano ridotti nei topi MeCP2y/-. Inoltre, il trattamento con Mirtazapina ha ristabilito la normale trasmissione eccitatoria (glutamatergica) e inibitoria (GABAergica) sia nella corteccia che nel midollo allungato dove risiedono i centri di controllo del respiro (Bittolo et al., 2016; Tabella 3). In conclusione, grazie ai suoi ridotti effetti collaterali e all’efficacia dimostrata nel modello animale della sindrome di Rett, la Mirtazapina si presenta come un ottimo candidato per il trattamento dei sintomi Rett, compresi i deficit respiratori e l’atrofia cerebrale, e merita di essere valutata in una sperimentazione clinica.
Tuttavia, per ottenere l’autorizzazione alla sperimentazione clinica di Mirtazapina nelle bambine Rett è necessario che siano soddisfatti i seguenti prerequisiti: 1) che sia dimostrata l’efficacia del trattamento anche nelle femmine di topo con mutazioni del gene MeCP2; 2) che si ottengano informazioni sul meccanismo attraverso cui Mirtazapina esercita i suoi effetti terapeutici nella sindrome di Rett.
Grazie al generoso contributo del-l’Associazione AIRETT abbiamo intrapreso uno studio per raccogliere le informazioni necessarie a redigere un dossier per ottenere l’autorizzazione ad un trial clinico di Fase 2 sull’efficacia della Mirtazapina nella sindrome Rett. In particolare, obiettivi specifici del progetto sono di: 1) verificare che nelle femmine eterozigoti MeCP2+/, la Mirtazapina produca gli stessi effetti osservati nei maschi sul ripristino dell’atrofia neuronale; 2) identificare i recettori e le vie di segnale intra- ed extra-cellulari attraverso cui Mirtazapina esercita i suoi effetti benefici sull’atrofia neuronale nei topi MeCP2.
In un primo esperimento è stata testata nelle femmine di 5-6 mesi una dose di 50 mg/kg che corrisponde a 5 volte la dose massima somministrabile negli esseri umani ed abbiamo notato una forte sedazione ed una diminuzione dei parametri con cui si misura la qualità della vita degli animali (phenotypic scoring). Abbiamo quindi ripetuto l’analisi con animali della stessa età, cioè 5-6 mesi, utilizzando una dose di 10 mg/kg che corrisponde alla dose massima somministrabile negli esseri umani e in questo caso abbiamo visto che la dose era ben tollerata e non abbiamo registrato effetti avversi. Il trattamento per un mese, a giorni alterni, non ha dimostrato miglioramenti a livello motorio come peraltro già in parte osservato preliminarmente nello studio sui topi maschi MeCP2y/- (Null) (Bittolo et al., 2016). Tuttavia abbiamo potuto osservare un miglioramento significativo delle aberrazioni del comportamento ansioso, confermando il risultato positivo riscontrato nei topi maschi (Bittolo et al., 2016). Infine, abbiamo osservato che Mirtazapina migliora la capacità delle femmine di costruire il nido, un comportamento caratteristico dei topi che richiede sia abilità di coordinamento motorio delle zampe anteriori, sia capacità cognitive e senso spaziale. La sperimentazione non è ancora conclusa e proseguirà con l’analisi anatomica dei cervelli dei topi femmina trattati per vedere se c’è stato un ripristino dell’atrofia cerebrale e una normalizzazione del fenotipo dei neuroni Glutammatergici e GABAergici.
Infine, dato che 6 mesi per un topo corrispondono circa a 35 anni per un essere umano, intendiamo ripetere il trattamento ad un’età dei topi corrispondente a circa 14-16 anni per gli umani, in modo da verificare se su un cervello più giovane Mirtazapina possa esplicare un maggiore effetto terapeutico.
Conclusioni.
Sebbene ancora non sia disponibile una cura per la sindrome di Rett, le ricerche degli ultimi anni hanno portato ad una migliore comprensione dei meccanismi patogenetici della sindrome di Rett consentendo di individuare alcuni bersagli su cui indirizzare le terapie. Sono stati quindi identificati dei potenziali farmaci che agiscono su recettori diversi innescando a livello cellulare varie tipologie di effetti. Alcuni farmaci promuovono il ripristino di una neurotrasmissione bilanciata tra segnali eccitatori e inibitori, altri una più efficiente produzione di energia cellulare che minimizzi la produzione di scorie ossidative dannose per la cellula, ed infine altri farmaci sono capaci di innescare i segnali dei fattori neurotrofici protettivi per la cellula. Significativamente, in base ai dati disponibili in letteratura, Mirtazapina potrebbe essere in grado di produrre tutti questi effetti contemporaneamente ed è per questo che la nostra sperimentazione su Mirtazapina continua.
Tabella1. Sperimentazione clinica nella sindrome di Rett CON ESITI POSITIVI oppure IN CORSO

Tabella 2. Sperimentazione clinica nella sindrome di Rett CON ESITI NEGATIVI

Tabella 3. Risultati principali della sperimentazione animale di Mirtazapina condotta all’Università di Trieste

Letteratura citata (in ordine di comparsa nel testo)
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. Jan 2007; 315(5815):1143-7.
Katz D, Bird A, Coenraads M, Gray SJ, Menon DU, Philpot DC, and Tarquinio DC. Rett Syndrome: Crossing the Threshold to Clinical Translation. Trends Neurosci. Feb 2016; 39(2): 100–113. - Szczesna K, de la Caridad O, Petazzi P, Soler M, Roa L, Saez MA, Fourcade S, Pujol A, Artuch-Iriberri R, Molero-Luis M, Vidal A, Huertas D, Esteller M. Improvement of the Rett syndrome phenotype in a MeCP2 mouse model upon treatment with levodopa and a dopa-decarboxylase inhibitor. Neuropsychopharmacology. 2014 Nov;39(12):2846-56.
- Nag N, Berger-Sweeney JE. Postnatal dietary choline supplementation alters behavior in a mouse model of Rett syndrome. Neurobiol Dis. 2007 May;26(2):473-80.
- Leoncini S, De Felice C, Signorini C, Zollo G, Cortelazzo A, Durand T, Galano JM, Guerranti R, Rossi M, Ciccoli L, Hayek J. Cytokine Dysregulation in MECP2- and CDKL5-Related Rett Syndrome: Relationships with Aberrant Redox Homeostasis, Inflammation, and ω-3 PUFAs. Oxid Med Cell Longev. 2015;2015:421624.
- Tang X, Kim J, Zhou L, Wengert E, Zhang L, Wu Z, Carromeu C, Muotri AR, Marchetto MC, Gage FH, Chen G. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc Natl Acad Sci U S A. 2016 Jan 19;113(3):751-6.
- Zoghbi HY, Percy AK, Glaze DG, Butler IJ, Riccardi VM. Reduction of biogenic amine levels in the Rett syndrome. N Engl J Med. 1985 Oct 10;313(15):921-4
Ide S, Itoh M, Goto Y. Defect in normal developmental increase of the brain biogenic amine concentrations in the mecp2-null mouse. Neurosci Lett. 2005 Sep 23;386(1):14-7. - Santos M, Summavielle T, Teixeira-Castro A, Silva-Fernandes A, Duarte-Silva S, Marques F, Martins L, Dierssen M, Oliveira P, Sousa N, Maciel P. Monoamine deficits in the brain of methyl-CpG binding protein 2 null mice suggest the involvement of the cerebral cortex in early stages of Rett syndrome. Neuroscience. 2010 Oct 13;170(2):453-67
Roux JC, Dura E, Moncla A, Mancini J, Villard L. Treatment with desipramine improves breathing and survival in a mouse model for Rett syndrome. Eur J Neurosci. 2007 Apr;25(7):1915-22. - Zanella S, Mebarek S, Lajard AM, Picard N, Dutschmann M, Hilaire G. Oral treatment with desipramine improves breathing and life span in Rett syndrome mouse model. Respir Physiol Neurobiol. 2008 Jan 1;160(1):116-21.
- Pacher P, Kecskemeti V. Cardiovascular side effects of new antidepressants and antipsychotics: new drugs, old concerns? Curr Pharm Des. 2004;10(20):2463-75.
- Cipriani A, Furukawa TA, Salanti G, Geddes JR, Higgins JP, Churchill R, Watanabe N, Nakagawa A, Omori IM, McGuire H, Tansella M, Barbui C. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009 Feb 28;373(9665):746-58.
- Burrows GD, Kremer CM. Mirtazapine: clinical advantages in the treatment of depression. J Clin Psychopharmacol. 1997 Apr;17 Suppl 1:34S-39S.
- Hartmann PM. Mirtazapine: a newer antidepressant. Am Fam Physician. 1999 Jan 1;59(1):159-61.
- Bittolo T, Raminelli CA, Deiana C, Baj G, Vaghi V, Ferrazzo S, Bernareggi A, and Tongiorgi E. Pharmacological treatment with mirtazapine rescues cortical atrophy and respiratory deficits in MeCP2 null mice. Sci. Rep. 2016 6, Article number: 19796.
- Katz DM, Berger-Sweeney JE, Eubanks JH, Justice MJ, Neul JL, Pozzo-Miller L, Blue ME, Christian D, Crawley JN, Giustetto M, Guy J, Howell CJ, Kron M, Nelson SB, Samaco RC, Schaevitz LR, St Hillaire-Clarke C, Young JL, Zoghbi HY, Mamounas LA. Preclinical research in Rett syndrome: setting the foundation for translational success. Dis Model Mech. 2012 Nov;5(6):733-45.
