Riportiamo l’articolo apparso su www.ilfattoquotidiano.it.
Segue il parere dell’Airett Research Team
da “Il Fatto Quotidiano – 4 gennaio 2016”
La ricerca sull’autismo ha fatto progressi nel corso del 2015. E a questi passi in avanti nella lotta contro una malattia, che solo in Italia colpisce circa 500mila persone, e per la quale le origini sono ancora in gran parte ignote, se ne aggiunge uno nuovo. Gli scienziati della Penn State University (Usa) hanno scoperto un nuovo bersaglio farmacologico in grado di salvaguardare dai deficit funzionali le cellule nervose umane derivate da pazienti con sindrome di Rett, grave forma di disturbo dello spettro autistico. La ricerca, guidata da Gong Chen, potrebbe portare a un nuovo trattamento per questa e altre manifestazioni della malattia. Un documento che descrive la ricerca sarà pubblicato sui ‘Proceedings of the National Academy of Sciences (Pnas).
“L’aspetto più emozionante di questa ricerca è che utilizza direttamente i neuroni umani provenienti da pazienti con sindrome di Rett come modello di malattia per indagare il meccanismo di base”, spiega Chen. “Pertanto, il nuovo bersaglio scoperto in questo studio potrebbe avere implicazioni cliniche dirette nel trattamento di questa patologia e potenzialmente di altri disturbi dello spettro autistico”).
I ricercatori hanno differenziato staminali derivate da cellule della pelle di pazienti affetti da sindrome di Rett in cellule nervose che possono essere studiate in laboratorio. Queste cellule nervose portano una mutazione nel gene Mecp2, ritenuta la causa della maggior parte dei casi di sindrome di Rett. I ricercatori hanno scoperto che in queste cellule nervose manca una molecola importante, Kcc2, che è fondamentale per la funzione delle cellule nervose normali e lo sviluppo del cervello. “Kcc2 controlla la funzione del neurotrasmettitore Gaba in un momento critico durante lo sviluppo cerebrale precoce”, evidenziano gli autori.
“È interessante notare che, quando abbiamo inserito questa molecola nei neuroni Rett, la funzione di Gaba è tornata alla normalità. Riteniamo quindi che aumentare la funzionalità di Kcc2 nei pazienti con sindrome di Rett possa portare a un potenziale nuovo trattamento“.
Il parere dell’Airett Research Team
KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome.
Tang X, Kim J, Zhou L, Wengert E, Zhang L, Wu Z, Carromeu C, Muotri AR, Marchetto MC, Gage FH, Chen G.
Proc Natl Acad Sci U S A. 2016 Jan 5. pii: 201524013
L’ articolo riportato da Quotidiano Sanità e pubblicato su PNAS è, a nostro avviso, interessante ma non estremamente innovativo. In breve, gli autori hanno studiato un modello cellulare di neuroni ottenuti a partire da fibroblasti di due pazienti, un maschio ed una femmina, (portatori di mutazioni mutazioni piuttosto rare di MeCP2) con l’obiettivo di comprendere perché nella sindrome di Rett si abbia un‘insorgenza ritardata dei sintomi.
Lo studio li ha portati ad evidenziare una diretta associazione tra una molecola, chiamata KCC2 e MECP2, il principale gene candidato nella Rett. KCC2 è molto importante perché esercita nella cellula nervosa una funzione di ripristino dallo stato di eccitazione ad inibizione (polarizzazione – depolarizzazione), controllando la funzione del neurotrasmettitore GABA ed è coinvolta in diverse patologie dello spettro autistico (Cellot et.al. Frontiers in Pediatrics, 2014), nella schizofrenia, nel dolore cronico e nella sindrome di Down. Secondo gli esperimenti qui riportati KCC2 risulta essere ridotto nei neuroni Rett. Tale riduzione altera il tradizionale ruolo inibitorio dei neurotrasmettitori GABA. Questo risultato non è nuovissimo perché già nel 2013 Mercè Pineda aveva riportato uno sbilanciamento nella concentrazione cerebrospinale di KCC2 e di NKCC1 in pazienti RTT (Duarte et.al. PLOSONE 2013).
Gli autori dimostrano inoltre che l’espressione di KCC2 è ridotta nello sviluppo cerebrale precoce, per poi diventare rilevante nell’adulto associando la diminuzione di KCC2 in conseguenza ad una mutazione di MECP2 al ritardato apparire dei sintomi nella bambine.
Molto importante è l’evidenza che il trattamento con IGF1 ripara il deficit di KCC2, quindi possiamo considerare questo studio una spiegazione all’efficacia di IGF1 nel migliorare la sintomatologia della sindrome di Rett. Si sottolinea come siano in elaborazione molecole che agiscono direttamente su KCC2 indipendentemente da Igf-1 e il cui effetto nella Rett potrebbe essere indagato. Per esempio per l’epilessia del neonato e per l’autismo vi sono stati dei trials con la bumetanide (farmaco già commercializzato) in Francia, con Y Ben Ari che è il maggiore esperto su questo potenziale trattamento.
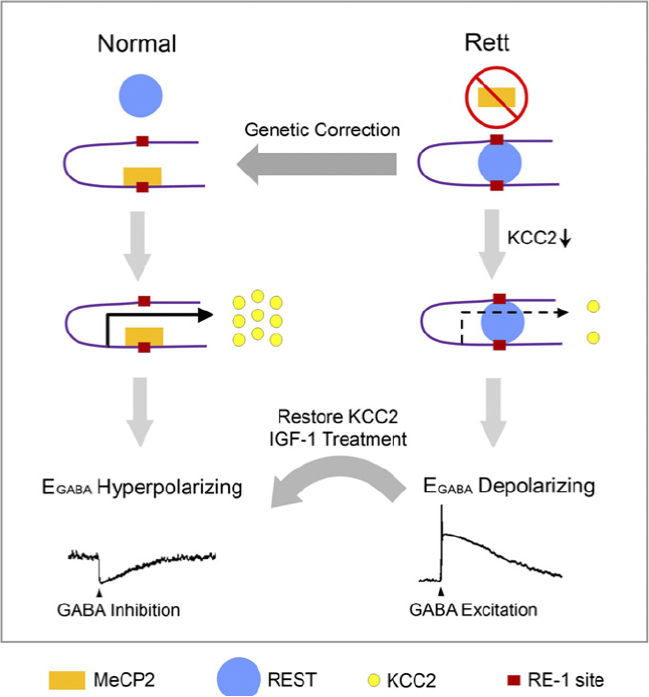 Nel lavoro è riportato un modello che illustra le relazioni tra MeCP2, KCC2 ed un fattore MECP2 regolerebbe KCC2 attraverso REST (un fattore trascrizionale che inibisce i geni neuronali), in un processo che è indipendente da IGF1. Osservando il grafico nella colonna a sinistra, si osserva come nei neuroni normali, MeCP2 (rettangolo giallo) può legarsi al sito di RE-1 (inibitore trascrizionale-quadratino rosso) all’interno del promotore KCC2 e impedendo il legame di REST Cerchio blu e lasciando che KCC2 venga prodotto nelle giuste quantità(cerchiolini gialli). Come evidenziato a destra, nei neuroni Rett, dove MeCP2 è carente, REST può legare con RE-1 sito nella regione del promotore KCC2 per sopprimere l’espressione KCC2. Ristabilendo una corretta concentrazione di MeCP2 o con il trattamento con IGF1 si può di nuovo ottenere una corretta espressione di KCC2.
Nel lavoro è riportato un modello che illustra le relazioni tra MeCP2, KCC2 ed un fattore MECP2 regolerebbe KCC2 attraverso REST (un fattore trascrizionale che inibisce i geni neuronali), in un processo che è indipendente da IGF1. Osservando il grafico nella colonna a sinistra, si osserva come nei neuroni normali, MeCP2 (rettangolo giallo) può legarsi al sito di RE-1 (inibitore trascrizionale-quadratino rosso) all’interno del promotore KCC2 e impedendo il legame di REST Cerchio blu e lasciando che KCC2 venga prodotto nelle giuste quantità(cerchiolini gialli). Come evidenziato a destra, nei neuroni Rett, dove MeCP2 è carente, REST può legare con RE-1 sito nella regione del promotore KCC2 per sopprimere l’espressione KCC2. Ristabilendo una corretta concentrazione di MeCP2 o con il trattamento con IGF1 si può di nuovo ottenere una corretta espressione di KCC2.
In conclusione il lavoro pur non indicando in realtà nessuna nuova terapia fa luce sui meccanismi alla base del funzionamento di IGF1 e su dove eventualmente indirizzare la ricerca per lo studio preclinico e clinico di farmaci già esistenti.
