
La presentazione si poneva due principali obiettivi:
a) illustrare a genitori e clinici le nuove tecnologie di sequenziamento (NGS sequenziamento di nuova generazione) in fase di attivazione presso l’Istituto Auxologico Italiano che consentiranno di fornire diagnosi più veloci, indagando contemporaneamente e più finemente, sia i geni noti per essere causativi della sindrome di Rett nella forma classica ed in quelle atipiche, sia i geni coinvolti nelle principali sindromi in diagnosi differenziale con la sindrome di Rett;
b) riferire i risultati preliminari di un progetto di ricerca in corso presso il nostro istituto con lo scopo di individuare nuovi geni causativi della sindrome di Rett, in quelle bambine che pur avendo un quadro clinico classico non presentano mutazioni nei geni noti.
a) Applicazione del sistema NGS all’attività di diagnostica della sindrome di Rett.
L’attività di diagnostica di una malattia genetica prevede lo studio di quei geni che si è dimostrato essere causa di una data sindrome.
Il principale gene causativo della sindrome di Rett è il gene MECP2, localizzato sul cromosoma X (Xq28) le cui mutazioni si riscontrano in circa il 95% delle bambine con la forma classica della sindrome di Rett e nel 50-70% di quelle con forma atipica.
Circa l’11-31% delle bambine con fenotipo Rett-like caratterizzato da insorgenza precoce di epilessia, il 28% delle bambine che associano epilessia ad insorgenza precoce e spasmi infantili ed il 9% di quelle che mostrano solo precoce insorgenza dell’epilessia hanno mutazioni nel gene CDKL5, anch’esso localizzato sul cromosoma X (Xp22).
Seppure con un frequenza di mutazione molto più limitata altri 2 geni sono stati recentemente inclusi tra i geni da considerare come putativi responsabili della sindrome di Rett: i) il gene FOXG1, localizzato sul cromosoma 14q12 per cui sono state descritte ad oggi 26 mutazioni puntiformi, 25 CNVs (Copy Number Variations), and 2 traslocazioni bilanciate t(2;14), è associato a forme congenite Rett-like, contraddistinte da epilessia precoce e da una severa microcefalia acquisita, ii) il gene MEF2C, sul cromosoma 5q14.3, individuato finora in pochi casi Rett-like, sempre in forme congenite con epilessia precoce e prevalenza di microduplicazioni.
Talvolta vi sono sindromi che condividono alcuni, ma non tutti i segni clinici essenziali. Vengono definite come sindromi in diagnosi differenziale. Talvolta la diagnosi clinica di una paziente appare controversa ed il medico dopo aver indagato i geni responsabili della sindrome in oggetto, che corrisponde principalmente al suo sospetto clinico, preferisce testare anche i geni responsabili delle patologie in diagnosi differenziale. Nel caso della sindrome di Rett, tra le principali diagnosi differenziali indicate in letteratura vi sono la sindrome di Angelman e la sindrome di Pitt-Hopkins, che condividono il riscontro frequente di alcuni segni clinici, quali il grave ritardo mentale, l’assenza o limitata presenza di linguaggio, la microcefalia acquisita e le crisi convulsive. Ciascuna delle 2 sindromi presenta ulteriori caratteristiche presenti anche nella Rett, ad esempio nella sindrome di Pitt-Hopkins sono descritte iperventilazione ed apnee, nella sindrome di Angelman scoliosi e disturbi del sonno. I difetti genetici della sindrome di Angelman includono mutazioni nel gene UBE3A che si indagano mediante sequenziamento, mentre i difetti genetici della sindrome di Pitt-Hopkins includono mutazioni nel gene TCF4.
Fino ad oggi il protocollo di sequenziamento prevedeva l’applicazione del sequenziamento diretto (sequenziamento di Sanger) oppure di un metodo di screening delle variazioni di sequenza, DHPLC, che richiede tempi di preparazione tecnica e di lettura della singola sequenza piuttosto lunghi. I tempi di analisi, senza tenere conto degli aspetti burocratici e delle liste di attesa, dall’arrivo del campione di sangue alla sequenza letta e refertata era di circa due settimane lavorative per un singolo gene di media lunghezza. Inoltre ogni singolo gene veniva sequenziato successivamente all’altro e per studiare 3 dei 4 geni principali e uno dei geni in diagnosi differenziale si impiegavano a volte anche sei mesi. Le metodiche di nuova generazione permettono un sequenziamento massivo capace di processare la sequenza di una quantità di basi del DNA molte maggiore in tempi velocissimi. Inoltre poiché tutti i frammenti saranno sequenziati potremmo riscontrare anche una maggiore accuratezza rispetto ai frammenti pretestati con metodo DHLPC, rivelando mutazioni che erano state perse. Tutti i geni di cui abbiamo parlato finora possono essere processati contemporaneamente nel giro di pochi giorni. Tuttavia non sarà possibile dare un risultato in tempi così brevi perché dovremo raccogliere un adeguato numero di pazienti e confermare con il metodo di sequenziamento tradizionale le mutazioni; si stima che quando la metodica sarà definitivamente a punto, in un tempo di 2 mesi si potranno fornire risposte per tutti i sei geni. Il sequenziamento NGS mantiene tuttavia il limite di non riuscire ad identificare delezioni o duplicazioni di interi esoni, per le quali resterà necessario completare lo studio diagnostico con una metodica dedicata a tale scopo, MLPA.
b) Ricerca di nuovi geni possibilmente causativi della sindrome di Rett
Una piccola percentuale di bambine con diagnosi clinica di sindrome di Rett, 5% con forma classica e un 40% circa con forme atipiche non hanno una diagnosi genetica, ossia non si conosce il gene responsabile della malattia. Allo scopo di identificare il difetto genetico anche in queste bambine, è stato proposto un progetto di ricerca cofinanziato dall’associazione sindrome di Rett, che prevede la collaborazione del nostro istituto e dei clinici afferenti al progetto, dott.ssa Vignoli dell’ospedale San Paolo, della dott.ssa Maria Pintaudi e prof.ssa Veneselli, dell’ospedale Gaslini, oltre che del dott. Giordano degli Spedali Civili di Brescia e della dott.ssa Ben Zeev di Tel Aviv. è stata selezionata una coorte di 23 bambine ed un maschio con fenotipo riconducibile alla forma classica della sindrome, 2 maschi e 6 bambine con encefalopatia epilettica precoce ed un caso familiare (per la presenza 2 sorelle con fenotipo Rett-like). Tutte le pazienti erano state precedentemente analizzate senza riscontro di difetti genetici per i geni MECP2, CDKL5 e FOXG1.
Il campione è stato processato in un esperimento di Whole Exome Sequencing (sequenziamento dell’intero esoma). Il termine esoma definisce quella parte del genoma, poco meno del 2%, contenente geni noti che producono proteine, e che si stima contenga l’85% dei geni responsabili di malattia. Il sequenziamento massivo dell’esoma permette almeno teoricamente di identificare tutte le variazioni di sequenza che un dato campione presenta se confrontate con le sequenze di riferimento (popolazioni di controllo) depositate nei databases. L’analisi dell’esoma viene eseguita sempre mediante tecnologia NGS, preparando librerie di DNA arricchite per la presenza di sequenze esomiche. Si ottengono così decine di migliaia di varianti che verranno analizzate con un approccio bioinformatico. L’analisi bioinformatica e la selezione delle varianti da considerare come patogenetiche rappresentano la parte più critica dello studio. Nella figura sottostante sono riportate le tappe dell’esperimento.
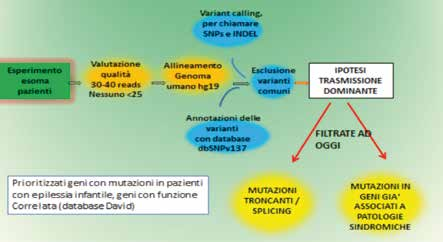
Dopo aver escluso le varianti comuni alla popolazione generale e perciò sicuramente non causa di malattia, abbiamo deciso come primo approccio in questo studio di ipotizzare una trasmissione di tipo dominante, de novo, ossia non trasmessa da genitori portatori. Abbiamo scelto varianti che corrispondessero a mutazioni di splicing o troncanti, ossia mutazioni che determinino la produzione di proteine non integre o mutazioni in geni già riportati associati a patologie sindromiche.
Ad oggi abbiamo selezionato 23 geni d’interesse, di cui sono state identificate 25 varianti che sono state riconfermate tutte con il metodo Sanger. Abbiamo indagato la presenza delle variazioni nelle coppie di genitori disponibili, verificando in 2 casi l’origine de novo ed in 12 la trasmissione parentale, escludendo così al momento il significato patogenetico di queste ultime varianti; nei rimanenti 9 casi lo studio della trasmissione parentale è in corso.
Confrontando i nostri risultati con un recentissimo lavoro di Veeramah (2013), che ha analizzato l’esoma in una decina di pazienti con encefalopatia epilettica, non abbiamo riscontrato nei nostri campioni le varianti identificate nei geni da lei indicati.
Tra i geni in cui abbiamo riscontrato tale varianti, vi sono alcuni geni già coinvolti in patologie sindromiche, quali EOAE (Early Onset Absence Epilepsies), Sintaxina Binding Protein associata a EIEE, Sodium Channel Protein Type 2 Subunit Alpha, Gamma-Aminobutyric Acid (Gaba) A Receptor, Subunit Delta, Gamma-Aminobutyric Acid (Gaba) A Receptor, Gamma 2 Associati A Dravet, Athrophin1, Dentatorubral Pallidoluysian Atrophy.
Non entrerò nei dettagli dei geni in cui sono state riscontrate varianti de novo, perché il dato è assolutamente preliminare. è in corso la validazione sul trascritto e dove possibile a livello funzionale di possibili varianti proteiche o proteine tronche, per dimostrare il ruolo causativo della malattia.
Le tappe successive dello studio prevedono la ricerca di candidati mediante ulteriori modelli di trasmissione. L’identificazione di nuovi candidati oltre a consentire la diagnosi ad un numero maggiore di pazienti permette di ampliare le conoscenze riguardo ai meccanismi inerenti la patogenesi della sindrome.
