
Convegno Lido di Camaiore 11 e 12 giugno 2005
 L’identificazione di un nuovo gene della SR apre nuove frontiere per la ricerca e fa ben sperare per il futuro, con un nuovo progetto che prevede l’impiego di cellule staminali mesenchimali.
L’identificazione di un nuovo gene della SR apre nuove frontiere per la ricerca e fa ben sperare per il futuro, con un nuovo progetto che prevede l’impiego di cellule staminali mesenchimali.
Professoressa Alessandra Renieri, Direttore della U.O.C. Genetica Medica – Università di Siena
Grazie ad uno studio collaborativo sulla sindrome di Rett è oggi disponibile uno strumento diagnostico addizionale per le pazienti con sindrome di Rett. E’ stato infatti identificato un nuovo gene responsabile della variante con convulsioni ad esordio precoce. Sono state descritte le nuove scoperte che ruotano intorno a questo gene. Durante l’intervento sono state inoltre affrontate altre tematiche, come il rischio di ricorrenza di coppie che hanno una figlia con la sindrome di Rett, ed il nuovo progetto sulla sindrome di Rett che è in fase di attuazione presso la Genetica Medica di Siena.
Rispondo con molto piacere all’invito della signora De Marchi di scrivere un piccolo riassunto su quanto affrontato durante la riunione annuale della vostra associazione a Lido di Camaiore l’11 Giugno 2005.
Già dal 1998, quando ancora non ero responsabile della U.O.C. Genetica Medica di Siena, ho iniziato ad interessarmi alle famiglie che avevano una bambina affetta da sindrome di Rett grazie alla stretta collaborazione con il reparto di Neuropsichiatria di Siena (prof. Michele Zappella e dott. Giuseppe Hayek).
Con il passare degli anni mi sono resa conto che sarebbe più corretto parlare delle “sindromi di Rett” piuttosto che della “sindrome di Rett” (Tabella 1).
| Tabella 1 |
|---|
| Le “sindromi di Rett” |
| Forma classica |
| Variante con recupero del linguaggio |
| Variante con insorgenza precoce delle convulsioni |
| Variante congenita |
| Variante «Forme fruste» |
| Variante con regressione tardiva |
Oltre alla forma classica, sono state infatti descritte almeno 5 varianti. Queste includono:
- la variante a linguaggio conservato in cui viene riacquisita dopo il periodo di regressione la capacità di espressione con brevi frasi;
- la variante con convulsioni ad esordio precoce che è caratterizzata da crisi convulsive che si manifestano prima del periodo di regressione;
- le “forme fruste” in cui i segni clinici caratteristici sono più sfumati;
- la variante congenita che è evidente sin dai primi mesi di vita;
- la variante a regressione tardiva.
Dal punto di vista genetico la sindrome di Rett è una malattia X-legata dominante, dovuta nella maggior parte dei casi a mutazioni “de novo” nel gene MECP2, localizzato sul cromosoma X, identificato nel 1999. Ad oggi solo in circa l’80% delle pazienti con diagnosi di sindrome di Rett classica è possibile mettere in evidenza una mutazione nel gene MECP2. Questa percentuale è molto più bassa se vengono considerate le varianti della sindrome. In questi 5 anni dalla scoperta del gene MECP2 sono stati fatti notevoli passi avanti nella comprensione dei processi biologici che portano alla sindrome di Rett. Tuttavia devono ancora essere chiariti molti aspetti.
Molti studi sono stati indirizzati e sono tuttora volti a comprendere il ruolo del gene MECP2 nella sindrome. Considerando l’organismo umano come una grande fabbrica, possiamo pensare ai geni/proteine come a degli operai. E’ stato dimostrato che il gene MECP2 è un regolatore della trascrizione ovvero un gene che coordina altri geni. Si può quindi considerare come un capo operaio in una grande fabbrica. Quali siano i geni regolati da MECP2 non è ancora del tutto chiaro. Il mio gruppo è stato uno dei protagonisti della scoperta di un nuovo gene della sindrome di Rett, il gene CDKL5. Abbiamo dimostrato che il gene CDKL5, se alterato, causa la variante con convulsioni ad esordio precoce della sindrome di Rett (Scala, E. et al. J Med Genet. 2005 Feb;42(2):103-7). Tale variante è stata per la prima volta descritta da Hanefeld nel 1985. Hanefeld ha descritto una bambina con spasmi infantili e ipsaritmia, che più tardi ha sviluppato la sindrome di Rett. L’insorgenza precoce di convulsioni (in alcuni casi spasmi infantili) è stata descritta in altre bambine che in seguito hanno sviluppato i segni caratteristici della sindrome di Rett (Goutières e Aicardi 1986; Maia et al. 1986). In questa variante non sono mai state riportate mutazioni del gene MECP2. Fino ad ora abbiamo identificato un’alterazione nel gene CDKL5 in 4 su 7 bambine con la variante con insorgenza precoce delle convulsioni. L’analisi del DNA di altre bambine con la sindrome di Rett classica e altre varianti non ha messo in evidenza alterazioni.
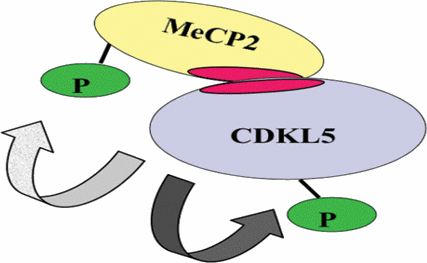
Una volta chiarito il legame tra CDKL5 e la sindrome di Rett abbiamo cercato di capire qualcosa di più sulla funzione di tale gene e come mai sia responsabile di un quadro clinico simile a quello causato da un’alterazione del gene MECP2. Le domande che ci siamo posti erano quindi: quali sono i compiti e le competenze dell’operaio CDKL5? Dal momento che la mancanza del capo operaio MECP2 e dell’operaio CDKL5 causa un effetto simile, i due operai lavorano probabilmente insieme, per lo stesso scopo? Grazie alla stretta collaborazione con il gruppo di Nicoletta Landsberger (Università dell’Insubria) e a quello di Vania Broccoli (Istituto San Raffaele di Milano) abbiamo iniziato a rispondere a queste domande (Fig.1) (Mari F et al. Hum Mol Genet. 2005 Jul 15;14(14):1935-46). Abbiamo dimostrato che la proteina CDKL5, come la proteina MeCP2, è espressa nel cervello, ovvero i due operai lavorano nello stesso settore della fabbrica. Inoltre abbiamo in parte chiarito qual è il compito dell’operaio CDKL5 e dimostrato che i due operai lavorano insieme, e che, in determinate circostanze, interagiscono direttamente. In particolare abbiamo dimostrato quali porzioni di CDKL5 e di MeCP2 sono importanti per la loro interazione. Inoltre abbiamo iniziato a capire che la funzione di CDKL5 è quella di aggiungere gruppi fosfato ad altre proteine tra le quali se stessa e MeCP2 (capacità di autofosforilazione e di fosforilazione).
Durante l’ampia discussione che è seguita alla breve relazione ho voluto affrontare un argomento che mi sta particolarmente a cuore. Si tratta del rischio di ricorrenza per le coppie che hanno già una bambina affetta da sindrome di Rett. La mutazione nel gene MECP2 (oppure nel gene CDKL5) insorge nel gamete di uno dei due genitori, il padre o la madre. Se la mutazione colpisce un gamete maturo (cellula uovo o spermatozoo) il rischio di ricorrenza per la coppia è zero. Nell’ovaio e nel testicolo ci sono però molte cellule che sono precursori delle cellule uovo e degli spermatozoi. Se la mutazione colpisce uno di questi precursori è possibile che più spermatozoi o più cellule uovo portino la mutazione. Se ciò accade due genitori sani possono generare più di una figlia affetta (mosaicismo germinale). Maggiore è l’entità del mosaicismo maggiore è il rischio di ricorrenza. L’entità del mosaicismo non è quantificabile con metodi di routine nelle singole coppie e quindi non è determinabile quale coppia ha una mutazione nel gamete maturo e quale coppia ha la mutazione nel precursore. Durante la consulenza genetica si fa presente alla coppia che il rischio di ricorrenza è molto basso ma non è zero. Per tale motivo si consiglia sempre di effettuare una diagnosi prenatale in ogni futura gravidanza tramite amniocentesi.
Prima di salutare le famiglie intervenute alla riunione ho voluto accennare ad un nuovo progetto di ricerca sulla sindrome di Rett che è avviato presso il nostro centro. L’avanzamento della ricerca sulla sindrome di Rett va a rilento perché il tessuto “malato” è inaccessibile. Abbiamo pensato di ovviare a questo ostacolo inducendo “in vitro” la differenziazione di cellule mesenchimali staminali in senso neuronale. Tale progetto, che è in corso di attuazione, prevede l’utilizzo di cellule staminali mesenchimali ottenute da prelievo di midollo osseo di pazienti con sindrome di Rett e di controlli sani (preferibilmente di un genitore o fratello/sorella). Sia le cellule dei pazienti che dei controlli verranno studiate durante il processo di differenziazione allo scopo di evidenziare eventuali differenze tra le cellule “malate” e quelle “sane”. Tale progetto ha finalità di ricerca, non è pertanto possibile definire se e quando i risultati che ne deriveranno potranno avere un impiego terapeutico per i pazienti sottoposti allo studio stesso (vedi il consenso informato per la partecipazione a questo progetto).
Le famiglie che fossero interessate a partecipare al progetto possono contattare direttamente la dott.ssa Francesca Mari, U.O.C. Genetica Medica, Università di Siena, Policlinico S. Maria alle Scotte, viale Bracci, 2, 53100 Siena, tel 0577233303, fax 0577233325, e-mail: mari@unisi.it.
