
L’intervento riassunto in queste pagine è in realtà il resoconto conclusivo del simposio parigino, e tuttavia ha il pregio di riassumere ciò di cui, nel corso della manifestazione, si è discusso nelle diverse giornate. Per questa ragione lo facciamo precedere alle altre relazioni, che i nostri lettori potranno approfondire nelle pagine seguenti.
Sintesi a cura della dott.ssa Silvia Russo – Istituto Auxologico di Milano
Dopo aver ringraziato gli organizzatori del Congresso Mondiale – Philippe Evrard, Gérard Nguyen, Helen Leonard, Leila Zribi, l’IRSF nella figura di Mary Joyce Griffin – la dottoressa Shanen ha dato un esaustivo riassunto della manifestazione che ha avuto il principale obiettivo di dare una panoramica dalle nuove acquisizioni molecolari ai trattamenti farmacologici mirati per la sindrome di Rett sviluppatesi a partire dalle conoscenze sempre più ampie sul ruolo di MECP2 nel sistema nervoso e del danno provocato dalla sua assenza sulle funzioni neuronali. MECP2 è il principale gene coinvolto nella sindrome di Rett, e gioca un ruolo complesso nel nucleo, con effetti diversi sulla trascrizione (Yasui et al, 2007, Chahrour, 2008). Basse concentrazioni di MECP2, determinano una perdita della regolazione genica dei geni target più rilevanti che sono Bdnf, Dlx5, Fxd1, IgFBP1, CRH, Cdkl5, e dei moltissimi altri che potrebbero essere controllati sia direttamente sia indirettamente da MECP2 ed essere tessuto o cellula specifici.
Per quanto riguarda lo sviluppo di possibilità terapeutiche, bisogna tener conto del fatto che non esiste un quadro clinico univoco per le pazienti Rett: lo studio della storia naturale e dell’EEG insieme ad altri parametri fisiologici potrebbe essere cruciale. Le pazienti hanno un background genetico ed XCI eterogenei, gli esperimenti in vitro su colture cellulari danno risultati discordanti, mentre i modelli murini restano l’approccio di studio più valido. Infatti il successo di una terapia si può valutare dall’osservazione diretta delle pazienti prima e dopo la somministrazione del farmaco e utilizzando modelli murini diversi. A tale proposito, è stata valutato, nei ceppi Mecp2-/Y , Mecp2308/Y il fenotipo neuronale, nei tre aspetti chimico, anatomico ed elettrofisiologico. Sono state riportate le anomalie dendritiche dei neuroni, e si è osservato che la morte dei neuroni non è la principale componente della patologia neurologica nella sindrome di Rett.
 Sindrome di Down
Sindrome di Down
Gli esperimenti di Bird sul modello murino Mecp2 lox-Stop, ossia topi in cui il gene endogeno Mecp2 è reso non funzionante dall’inserzione di una sequenza, lox-Stop appunto, che impedisce l’espressione del gene, hanno dimostrato che ripristinando l’espressione di Mecp2 si ottiene un miglioramento del quadro clinico e la reversione dei sintomi.
D’altro canto vi sono studi su topi che esprimono una doppia dose di Mecp2 rispetto al wt, sviluppano i sintomi neurologici, seppure con un’insorgenza più tardiva.
 Sindrome di Rett
Sindrome di Rett
Le possibilità terapeutiche si possono così riassumere:
- strategie di riparazione
- a livello del gene
- mediante reintroduzione della proteine
- strategie per modulare l’espressione del gene e quindi della proteina mediante
- Upregolazione delle cellule normali nelle eterozigoti
- Riattivazione del gene normale
- riparazione post trascrizionale
- Trattamento con un farmaco che permette la lettura del codice genico senza fermarsi ai codoni di stop, finora sperimentato nella fibrosi cistica e nella distrofia muscolare
- Trattamenti mirati all’attività di Mecp2 nei neuroni:
- Per valutare se si può migliorare la funzione proteica e se un’altra proteina può prendere il sopravvento.
Entrando nel dettaglio la prima distinzione quando si parla di terapia per una malattia genetica avviene tra terapia a livello del gene, intesa come a) terapia genica o b) riparazione post-trascrizionale, e terapia a livello della proteina, che comporta la reintroduzione della proteina o un’azione sui geni modificatori che vengono alterati dalla presenza di un gene MeCP2 mutato.
La TERAPIA GENICA ha qualche possibilità di successo in patologie monogeniche, in cui il gene causativo è noto e se i tessuti malati sono accessibili.
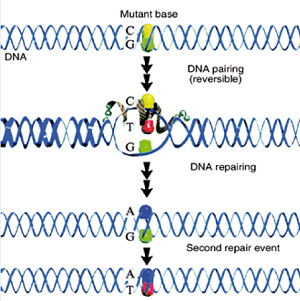
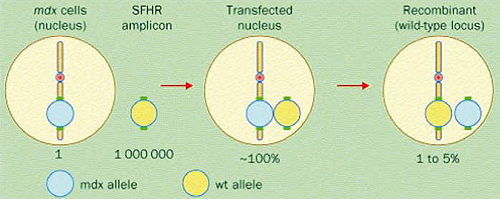
La riparazione dell’allele mutato si esegue introducendo un oligonucleotide modificato sulla singola elica che poi viene replicata inserendo l’allele normale; rimane tuttavia la questione efficienza che varia dall’1 al 5%.
Un approccio alternativo consiste nell’agire sull’inattivazione casuale del cromosoma X determinando preferenzialmente l’attivazione dell’allele normale.
La riparazione post-trascrizionale può consistere nell’utilizzo di aminoglisidi, molecole che permettono di oltrepassare i codoni di stop e produrre quindi la proteina MECP2. Questa metodica ha evidenziato alcuni problemi quali il fatto che è limitata alle sole mutazioni di stop, è tossica e può introdurre sostituzioni aminoacidiche al sito di stop. Recentemente è stata sviluppata una nuova molecola PTC124, che sfruttando la stessa strategia, avrebbe alcuni importanti vantaggi. E’ somministrabile oralmente, ha una bassa tossicità ed è stata testata per la fibrosi cistica e per la Distrofia Muscolare di Duchenne con risultati molto promettenti. Nella seconda fase del trial, i bambini con DMD avevano migliorato la loro deambulazione e nella fibrosi cistica la conduttanza al cloro.
A livello della proteina sono stati riportati alcuni studi non ancora pubblicati. Laccone mostra esperimenti di reintroduzione, mediante iniezione intraperitoneale, di una miscela della proteina di fusione con entrambe le isoforme proteiche TAT-MeCP2e1 ed TAT-MeCP2e2 nel topo Mecp2-/Y.
La proteina sembrerebbe oltrepassare la barriera emato-encefalica e giungere ai neuroni cerebrali. Si osservano come conseguenze, la reversione dell’iperacetilazione di H3, una maggior durata della vita e un miglioramento delle abilità motorie.
Il trial eseguito con Betaina e folati su un campione di 73 pazienti Rett, riferito da Percy e Glaze non avrebbe determinato variazioni su parametri oggettivamente misurabili, ma solo un miglioramento riferito dai genitori.
E’ stata osservata un’eterogeneità cellulare nel livello di espressione di Mecp2 in cervelli normali, con sottopopolazioni di cellule che esibiscono un’elevata espressione di MeCP2 (MeCP2 hi) e rimanenti sottopopolazioni che mostrano un’espressione ridotta (MeCP2lo), nelle femmine RTT eterozigoti l’espressione di MeCP2 è più ridotta anche nelle cellule wild type.
Sarebbe importante identificare strategie per potenziare l’espressione di MeCP2 wt.
A questo proposito viene riportato lo studio di Cassell che dimostrava come dopo la somministrazione per 10 giorni consecutivi di fluoxetina o di cocaina in un campione di ratti adulti l’espressione di MECP2 e di MBD1, incrementava coinvolgendo prevalentemente il sistema serotoninergico. I potenziali risultati terapeutici provengono dai laboratori dell’Eubanks circa l’incremento di espressione nelle femmine eterozigoti.
Studi sul modello Mecp2 308/Y provano che elevati livelli serici di corticosterone sono responsabili del comportamento ansioso e della risposta alterata allo stress, e pongono la questione della necessità di trial clinici sui pazienti per la somministrazione di farmaci ansiolitici.
L’approccio neurobiologico ha evidenziato che nei topi il deficit di Mecp2 è associato a limitati livelli di BDNF, mentre una maggiore espressione di BDNF ha effetti migliorativi. Il trattamento con ampakina aumenta l’espressione di Mecp2 e migliora le funzioni respiratorie nel topo Mecp2-/yJaenisch.
Shanen riferisce infine una serie di trattamenti sperimentati finora nei modelli murini:
- Colina sembra migliorare la coordinazione motoria e riduce l’ansia nei topi Mecp2-/yJaenisch
- Cerebrolisina in Mecp2308/y ha effetti sul comportamento e sull’aspetto neuropatologico
- Desipramina migliora la sopravvivenza e le funzioni respiratorie, ma non le difficoltà di accrescimento, la microcefalia e la limitata capacità motoria.
Con la desipramina è in corso un trial sulle pazienti, di cui si conoscono gli effetti collaterali, la tossicità cardiaca e gli effetti anticolinergici.
Un ruolo importante è anche quello dell’arricchimento ambientale; è stato dimostrato sui modelli murini che migliori molto la coordinazione motoria nelle femmine eterozigoti.
Per concludere, la terapia genica è ancora una strada futura, mentre le recenti acquisizioni sul funzionamento del sistema nervoso centrale aprono le porte a nuove terapie nell’immediato futuro, riproponendo farmaci esistenti, sviluppando nuovi farmaci e rilanciando l’importanza della stimolazione ambientale. E’ necessario sperimentare trials sui topi femmine e pianificare trials clinici per agenti selezionati.
