
Maurizio Giustetto – Silvia Russo
Dipartimento di Neuroscienze e Istituto Nazionale di Neuroscienze, Università di Torino

La sera del 24 giugno è stato inaugurato il “Basic Research Symposium” che è stato organizzato e diretto dalle proff.sse G. Mandel (Oregon, USA) e H.Y. Zoghbi (Texas, USA).
Come riportato durante l’intervento inaugurale dalle organizzatrici di questo Simposio, negli ultimi anni hanno abbiamo assistito a grandi cambiamenti nel modo in cui pensiamo a questa malattia. Nonostante le basi neurobiologiche della malattia stiano diventando sempre più chiare, rimangono ancora domande fondamentali a cui rispondere, come ad esempio quale sia il vero ruolo della proteina MeCP2 nello sviluppo e nel funzionamento del sistema nervoso centrale e quali sono le pathways (specifici circuiti di molecole che interagiscono tra di loro) che sono alterate dalla patologia. Solo acquisendo continuamente nuove conoscenze sperimentali, hanno concluso le due ricercatrici, sarà possibile da parte del mondo scientifico risolvere l’eziologia cellulare e molecolare della patologia Rett e testare sempre nuove cure farmacologiche o terapie innovative.
La prima sessione di questo simposio era dedicata agli aspetti neurobiologici di base del ruolo della proteina MeCP2 ed ha visto come primo relatore colui che ne ha scoperto la sua funzione, il dott. A.Bird (Edinburgh, UK). Dopo avere ricapitolato i molteplici aspetti sulle recenti scoperte dedicate alla funzione neuronale di MeCP2, il dott. Bird ha esposto nuovi dati che riguardano l’interazione tra MeCP2 ed il complesso NCoR/SMRT nella regolazione dell’espressione di nuovi geni (fig.1). Secondo i dati presentati, la mutazione di MeCP2 andrebbe a perturbare quest’interazione scaturendo così in un processo patologico che potrebbe essere fondamentale per l’insorgere della sindrome di Rett.
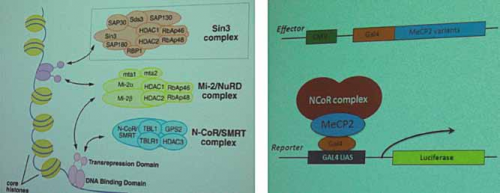 Fig. 1 – I complessi proteici di corepressori, come NCoR, contengono le iston-deacetilasi (HDAC) e vengono poi reclutati dalle proteine che legano il DNA. MeCP2 è un mediatore globale della repressione trascrizionale dipendente dalle HDAC, funge da ponte legandosi da un lato al DNA e dall’altro al complesso proteico.
Fig. 1 – I complessi proteici di corepressori, come NCoR, contengono le iston-deacetilasi (HDAC) e vengono poi reclutati dalle proteine che legano il DNA. MeCP2 è un mediatore globale della repressione trascrizionale dipendente dalle HDAC, funge da ponte legandosi da un lato al DNA e dall’altro al complesso proteico.
Il secondo intervento è stato presentato dal dott. D. Ebert (del gruppo del prof. Greenberg, Boston, USA) collegandosi a quanto appena descritto da A. Bird. Il dott. Ebert ha mostrato infatti i meccanismi molecolari fini attraverso cui la proteina MeCP2 è in grado di legarsi al complesso di NCoR per modulare la trascrizione e quali potrebbero essere le cause dell’alterazione di tale legame. Gli studi mostrati indicano che la corretta fosforilazione sull’aminoacido treonina 308 di MeCP2 possa abolire l’interazione della proteina con il complesso NCoR-HDAC3 danneggiando la capacità di MeCP2 di funzionare da repressore trascrizionale giocando un ruolo fondamentale per la patologia. Si portano evidenze che la fosforilazione della T308 regoli le arborizzazioni dendritiche in neuroni piramidali dell’ippocampo.
Nella sindrome di Rett, nonostante il difetto genetico sia presente già nelle cellule germinali, la malattie esordisce nei primi anni di vita dopo un periodo di normalità, con peculiare stadio di regressione. La motivazione di ciò non è ancora chiara. Partendo da un precedente studio che mostrava come se nel topo adulto si impediva la produzione di MeCP2 si aveva la malattia, la dott.ssa Ballas ha creato due modelli di topo che differiscono per il momento in cui viene inibita la produzione della proteina MeCP2. In un topo a) MeCP2 smette di essere prodotto a 5 settimane di vita, periodo che corrisponde all’insorgenza della regressione e primi sintomi, e b) nel secondo smette di essere prodotto a 10 settimane, età considerata adulta. Sono stati studiati modelli animali di entrambi i sessi. Si è riscontrata un’identica evoluzione della malattia, con una differenza tra i maschi che sviluppano immediatamente la sindrome appena MeCP2 viene a mancare e le femmine che la sviluppano alla 20 settimana come avviene nelle mutazioni germinali.
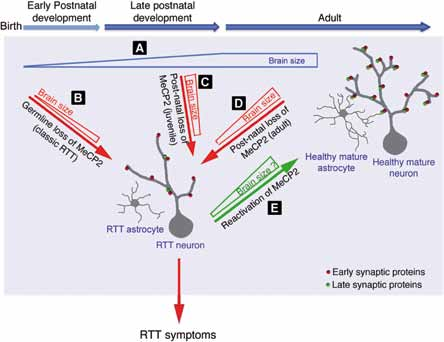 Fig. 2 – Modello della funzione di MeCP2 durante il periodo critico dello sviluppo postnatale del cervello e nel cervello maturo.
Fig. 2 – Modello della funzione di MeCP2 durante il periodo critico dello sviluppo postnatale del cervello e nel cervello maturo.
Lo studio dimostra (Fig. 2) come, venendo a mancare MeCP2 sia nell’età tardo giovanile sia nell’età adulta, la malattia evolve in modo simile a quando MeCP2 manca da sempre. Nel modello sano il cervello continuerebbe a crescere dopo la nascita e i neuroni raggiungerebbero una maggiore complessità, processo che coinvolge l’interazione di 2 tipi di cellule, i neuroni e la glia. In assenza di MeCP2 sia nelle cellule germinali, sia a partire da periodi successivi, i neuroni e gli astrociti assumono una struttura semplificata, non crescono e la massa del cervello appare restringersi con una maggiore densità cellulare. L’osservazione che la mancata funzione di MeCP2 dall’embrione, piuttosto che in epoca giovanile o adulta conferiscano sempre un sindrome di Rett classica, fanno presupporre che la regressione che si osserva nelle bambine Rett, sia conseguenza di una reale riduzione delle caratteristiche funzionali ed anatomiche del cervello. Queste conoscenze sono importanti perché fanno luce sulla plasticità di questo periodo e sulle possibilità di revertire i sintomi della malattia. Lo studio ha inoltre evidenziato che il numero di specifiche proteine sinaptiche è ridotto, un risultato che potrebbe indirizzare verso nuovi potenziali target per l’intervento terapeutico.
Nell’intervento successivo la dottoressa Donohoe del Burke Medical Research Institute, si basa sulla osservazione che la proteina MeCP2 condivide molte caratteristiche con gli istoni (proteine che interagiscono direttamente con il filamento di DNA intercalandosi con una geometria molto precisa), è ricca di aminoacidi che possono essere modificati dopo che la proteina è stata prodotta (modificazioni post-traduzionali, PMT), ma a parte l’effetto della fosforilazione che può variare la sua capacità di legarsi al DNA, non erano state identificate ad oggi molecole capaci di modificazioni post-traduzionali di MeCP2. La dott Dohonoe ha indagato se un particolare gruppo di proteine SUMO (small ubiquitin modifiers) capaci di PMT e coinvolte nell’interazione delle molecole con il DNA e nel suo riparo, interagissero con MeCP2 e in che modo. Lo studio della dott.ssa Donhoe ha dimostrato che questa interazione esiste, è dipendente dall’attività neurale ed è mediata da una molecola RanBP2. Quindi MeCP2 viene sumoilato e modifica la sua attività trascrizionale. In particolare questo permette di aggiungere qualche tassello alla relazione tra MeCP2 e un gruppo di molecole (L1, long interspersed nuclear element 1) che sono importanti durante lo sviluppo del cervello e influenzano l’espressione genica e le funzioni neuronali.
Nell’intervento successivo la dott.ssa Shatz della Stantford University affronta un argomento piuttosto nuovo, ossia la relazione tra plasticità neuronale e sistema immunitario. Il sistema immunitario e quello nervoso possono condividere lo stesso linguaggio. Le molecole che agiscono sul sistema immunitario, MHC di classe I, sono presenti nel sistema nervoso, come dimostrato in passato dalla stessa Shatz, e possono influenzare la capacità dei neuroni di evolvere in seguito a nuove esperienze. è possibile che l’espressione di MHCI sia aumentata nel topo MeCP2 difettivo e che immunitario questo tipo di alterazione molecolare durante lo sviluppo possa avere dirette conseguenze sulla maturazione delle sinapsi.
Nella sessione sulle basi neurobiologiche della sindrome, durante il suo intervento il Dr. Heintz riferisce sull’importanza della sesta base, idrossimetilcitosina. Quando si parla di metilazione del DNA ci si riferisce alle Citosine (una delle basi del DNA) metilate (metilcitosine), esiste però una variante, appunto l’idrossimetilcitosina, ed il rapporto tra idrossimetilcitosina e m-citosina fornisce una misura dell’accessibilità del DNA stesso. Secondo i dati di Heintz, MeCP2 interagisce direttamente con le idrossimetilcitosine, legandosi all’interno dei geni neuronali di cui regola la trascrizione. Sempre parlando di metilazione, il dott. Song riporta esperimenti di sequenziamento di tutte le regioni metilate del DNA con un approccio di nuova generazione che sequenzia base per base, rivelando il ruolo della metilazione non CpG nel cervello del mammifero adulto. L’osservazione importante di questo studio è che la metilazione di specifici geni neuronali viene modificata dall’attività.
In un altro interessante intervento, il dott. M. Green (Boston, USA) ha mostrato le potenzialità terapeutiche di alcune sostanze che sarebbero in grado di riattivare l’espressione dell’allele sano del gene MeCP2, allele che in circa il 50% delle cellule nelle femmine eterozigoti viene a trovarsi in un naturale stato di silenziamento. Con il suo studio identifica 13 molecole non prodotte da geni sul cromosoma X che contribuiscono al processo di inattivazione del cromosoma X e riporta esperimenti per riattivare in modelli cellulari di topo e uomo, il gene Mecp2.
La dott.ssa K. Krishnan del laboratorio del dott. J. Huang (Cold Spring Harbor, USA) ha mostrato che i circuiti corticali che sottendono alle funzioni visive maturano più in fretta negli animali Mecp2-KO e ne ha spiegato nei dettagli le cause alla base. Se questo tipo di difetto nello sviluppo delle connessioni nervose fosse esteso a tutte le aree corticali, questo potrebbe spiegare le alterazioni di tipo sensoriale associate alla patologia.
Nella giornata di martedì 26, dedicata alle scoperte più nuove provenienti dalla ricerca sulla sindrome di Rett, la prima presentazione delle dott.ssa Abdala Sheik era incentrata sul problema delle apnee che possono ridurre la saturazione dell’ossigeno nelle pazienti a livello patologico. Sono stati studiati modelli di topo Mecp2 +/-, femmine quindi, esplorando l’ipotesi che le aritmie del respiro abbiano inizio in una regione precisa, il nucleo Kolliker-Fuse (KF), coinvolto nel controllo della fase post-inspiratoria, quella che è alterata nelle pazienti Rett. Partendo dall’ipotesi che l’ini-
bizione del KF dipendesse da un’insufficienza dei recettori GABA e fosse la causa delle anomalie respiratorie nelle pazienti Rett, hanno fatto risalire i valori di GABA nelle femmine di topo Rett e in effetti le anomalie respiratorie miglioravano; viceversa bloccando i recettori GABAA in animali normali le apnee comparivano. Questo risultato dimostra che il deficit respiratorio nella Rett non deriva da una permanente distruzione del sistema nervoso centrale e apre spiragli per la cura di questo importante sintomo.
Nella comunicazione successiva il Dr. Kpinis riporta l’aggiornamento dei risultati recentemente pubblicati ed in parte riportati sul sito di AIRETT, che evidenziano come le cellule della glia ed alcune cellule della linea mieloide siano importanti nello sviluppo della sindrome di Rett. Il trapianto di midollo nel modello di topo Mecp2 difettivo sia in maschi sia in femmine determina un sostanziale miglioramento dei sintomi della malattia.
